Table of Contents
Filed Pursuant to Rule 424(b)(5)
Registration Statement No. 333-238991
Prospectus Supplement
(To Prospectus dated June 16, 2020)
4,629,630 Shares

NGM Biopharmaceuticals, Inc.
Common Stock
We are offering 4,629,630 shares of our common stock pursuant to this prospectus supplement. Our common stock is listed on The Nasdaq Global Select Market under the trading symbol “NGM.” On January 5, 2021, the last reported sale price of our common stock on The Nasdaq Global Select Market was $27.99 per share.
Investing in our common stock involves a high degree of risk. See “Risk Factors” beginning on page S-14 of this prospectus supplement.
| Per Share | Total | |||||||
| Public offering price |
$ | 27.00 | $ | 125,000,010 | ||||
| Underwriting discounts and commissions(1) |
$ | 1.62 | $ | 7,500,001 | ||||
| Proceeds, before expenses, to us |
$ | 25.38 | $ | 117,500,009 | ||||
| (1) | See “Underwriting” for additional disclosure regarding underwriting discounts and commissions and estimated expenses. |
We have granted the underwriters an option to purchase up to an additional 694,444 shares of our common stock within 30 days from the date of this prospectus supplement.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus supplement or the accompanying prospectus. Any representation to the contrary is a criminal offense.
The underwriters expect to deliver the shares of common stock against payment on or about January 8, 2021.
| Goldman Sachs & Co. LLC | Citigroup | Cowen |
| Raymond James | B. Riley Securities | |
Prospectus supplement dated January 5, 2021
Table of Contents
PROSPECTUS SUPPLEMENT
| Page | ||||
| S-1 | ||||
| S-14 | ||||
| S-71 | ||||
| S-73 | ||||
| S-74 | ||||
| S-76 | ||||
| Material U.S. Federal Income Tax Consequences to Non-U.S. Holders |
S-77 | |||
| S-81 | ||||
| S-88 | ||||
| S-88 | ||||
| S-88 | ||||
| S-89 | ||||
PROSPECTUS
| 1 | ||||
| 5 | ||||
| 6 | ||||
| 8 | ||||
| 9 | ||||
| 14 | ||||
| 20 | ||||
| 22 | ||||
| 25 | ||||
| 27 | ||||
| 27 | ||||
| 27 | ||||
| 27 |
S-i
Table of Contents
We have not, and the underwriters have not, authorized anyone to provide you with any information or to make any representation, other than those contained or incorporated by reference in this prospectus supplement and the accompanying prospectus, which together we sometimes refer to generally as the prospectus, or in any free writing prospectus prepared by us or on our behalf or to which we have referred you. Neither we nor any of the underwriters take any responsibility for, and provide no assurance as to the reliability of, any other information that others may give you. This prospectus supplement and the accompanying prospectus are an offer to sell only the shares of our common stock offered hereby, but only in circumstances and in jurisdictions where it is lawful to so do. The information contained or incorporated by reference in this prospectus supplement and the accompanying prospectus is accurate only as of its date, regardless of the time of delivery of this prospectus supplement and the accompanying prospectus or of any sale of our common stock.
For investors outside the United States: neither we nor any of the underwriters have done anything that would permit this offering or the possession or distribution of this prospectus supplement, the accompanying prospectus or any free writing prospectus that we have authorized for use in connection with this offering in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus supplement, the accompanying prospectus and any free writing prospectus that we have authorized for use in connection with this offering must inform themselves about, and observe any restrictions relating to, this offering and the distribution of this prospectus supplement, the accompanying prospectus and any free writing prospectus that we have authorized for use in connection with this offering outside the United States.
About This Prospectus Supplement
This document consists of two parts. The first part is this prospectus supplement, which describes the terms of this offering of our common stock and also adds to and updates information contained in the accompanying prospectus and the documents incorporated by reference in this prospectus supplement. The second part is the accompanying prospectus dated June 16, 2020, which includes the documents incorporated by reference therein and provides more general information. To the extent the information contained in this prospectus supplement differs or varies from the information contained in the accompanying prospectus or the documents incorporated by reference herein or therein, you should rely on the information in this prospectus supplement. Generally, when we refer to the prospectus, we are referring to this prospectus supplement and the accompanying prospectus combined. You should read both this prospectus supplement and the accompanying prospectus, together with the documents incorporated by reference herein and therein and any additional information described under the heading “Where You Can Find More Information.”
S-ii
Table of Contents
This summary highlights selected information appearing elsewhere or incorporated by reference in this prospectus supplement and the accompanying prospectus and does not contain all of the information that you need to consider in making your investment decision. This prospectus supplement and the accompanying prospectus include information about the shares of common stock that we are offering as well as information regarding our business. You should read this prospectus supplement and the accompanying prospectus, including the information incorporated by reference and any free writing prospectus that we have authorized for use in connection with this offering, in their entirety. You should also carefully consider the information set forth under “Risk Factors” beginning on page S-14 of this prospectus supplement before making your investment decision.
Overview
We are a biopharmaceutical company focused on discovering and developing novel therapeutics based on scientific understanding of key biological pathways underlying liver and metabolic diseases, retinal diseases and cancer. These diseases represent a significant burden for healthcare systems and, in some cases, are leading causes of morbidity and mortality. Since the commencement of our operations in 2008, we have generated a robust portfolio of product candidates. Our six most advanced product candidates are presented below:
![]()
NASH = non-alcoholic steatohepatitis; FGF = fibroblast growth factor; GDF = growth differentiation factor; KLB = beta-klotho; C3 = Component 3; GFRAL = glial cell-derived neurotrophic factor receptor alpha-like; CACS = Cancer Anorexia/Cachexia Syndrome; ILT2 = Immunoglobulin-like transcript 2; ILT4 = Immunoglobulin-like transcript 4; LAIR1 = Leukocyte-associated immunoglobulin-like receptor 1
In 2015, we entered into a research collaboration, product development and license agreement, or the Collaboration Agreement, with Merck Sharp & Dohme Corp., or Merck, that allows us to develop multiple product candidates in parallel without bearing substantially greater costs or incurring significantly greater risk compared to developing product candidates, such as aldafermin (formerly NGM282), an engineered version of the human hormone fibroblast growth factor 19, or FGF19, on our
S-1
Table of Contents
own. In March 2019, Merck exercised its option to extend the research phase of the collaboration through March 16, 2022 and has the unilateral right to extend it again through March 16, 2024. As part of the extension through March 16, 2022, Merck agreed to continue to fund our research and development efforts up to $75.0 million each year consistent with the initial five-year term and, in lieu of a $20.0 million extension fee payable to us, Merck agreed to make additional payments totaling up to $20.0 million in support of our research and development activities across 2021 and the first quarter of 2022.
Merck generally has a one-time right to exercise its option to an exclusive, worldwide license when a program completes a human proof-of-concept trial. In November 2018, Merck exercised its option to license MK-3655 (formerly NGM313), an agonistic antibody selectively activating fibroblast growth factor receptor 1c/beta-klotho, or FGFR1c/KLB, which is a potential treatment for non-alcoholic steatohepatitis, or NASH. The collaboration enables us to develop more product candidates for major indications than we could likely advance on our own. We retain an option, when a product candidate has advanced to Phase 3 clinical trials, to participate in up to 50% of the economic return from that product candidate if it becomes an approved medicine by agreeing to share up to 50% of the costs of development. If we do not elect this option, we will instead receive milestone and royalty payments and we will not be required to share in development costs. Overall, the Merck collaboration provides us with robust research and development support, while allowing us to retain our research independence and the option to split costs and profits on individual product candidates Merck elects to advance.
In 2020, we suspended development activities related to multiple product candidates for portfolio management considerations in order to concentrate our resources on aldafermin and certain other product candidates subject to our Merck collaboration. Most recently, in December 2020, based on the overall clinical experience with both NGM386 (a once-daily growth differentiating factor 15, or GDF15, agonist product candidate we suspended development of earlier in 2020) and NGM395 (a long-acting GDF15 agonist product candidate), we decided to suspend development of the NGM395 program after we have completed the ongoing Phase 1 clinical trial evaluating safety, tolerability and pharmacokinetics, or PK, in obese but otherwise healthy adults. We remain interested in the potential applications of a GDF15 agonist, but we believe antagonizing the GDF15 receptor, glial-cell derived neurotropin factor receptor alpha-like, or GFRAL, with NGM120, one of our product candidates described below, in patients with cancer has a stronger near-term rationale for development.
All of our product candidates other than aldafermin, NGM395 and NGM386 are subject to our Merck collaboration.
Aldafermin in NASH
Aldafermin, an engineered version of the human hormone FGF19 that is administered through a once-daily subcutaneous injection, has demonstrated the ability to rapidly improve NASH and reverse liver fibrosis in clinical and preclinical studies. We believe the combination of breadth, magnitude and speed of effect demonstrated by aldafermin in these studies results in an agent that, if ultimately approved, could provide a needed medicine for physicians to treat NASH patients with moderate to advanced fibrosis. Aldafermin is currently being tested in the ongoing Phase 2b ALPINE 2/3 trial in patients with NASH with stage 2 and 3, or F2-F3, liver fibrosis, which completed enrollment in September 2020, and in the ongoing ALPINE 4 trial in patients with NASH with stage 4, or F4, liver fibrosis and well-compensated cirrhosis, which commenced enrollment in February 2020. We expect to report topline data from the ALPINE 2/3 trial in the second quarter of 2021. Aldafermin was excluded from the Merck collaboration at the inception of the collaboration and remains wholly-owned by us.
S-2
Table of Contents
In August 2020, we announced final data from our completed double-blind, randomized, placebo-controlled Phase 2 clinical study (Cohort 4) of aldafermin, which enrolled patients with biopsy-confirmed NASH with F2-F3 liver fibrosis, including a new analysis of Cohort 4 data from NASH patients with F3 liver fibrosis. Cohort 4 was the final reported cohort from our adaptive Phase 2 clinical study of aldafermin in NASH.
Cohort 4 was statistically powered to demonstrate the effect of 1 mg aldafermin treatment versus placebo on the primary endpoint of change in absolute liver fat content, or LFC, which achieved statistical significance. In addition, the study assessed secondary and exploratory endpoints of liver histology and biomarkers of disease activity. The histology results revealed that treatment with aldafermin led to clinically meaningful improvements at 24 weeks versus placebo in fibrosis improvement of ³1 stage with no worsening of NASH (38% of aldafermin-treated patients vs. 18% placebo) and in resolution of NASH with no worsening of liver fibrosis (24% of aldafermin-treated patients vs. 9% placebo). The study also demonstrated a statistically significant impact on the combined endpoint of both fibrosis improvement and resolution of NASH (22% in aldafermin-treated patients vs. 0% placebo), which the FDA has indicated in draft industry guidance may be an acceptable development pathway.
| Summary of Cohort 4 Histology Data1 | ||||||||
| Proportion of Patients Achieving Endpoints |
Aldafermin 1 mg (n=50) |
Placebo (n=22) |
||||||
| Fibrosis improvement (>1 stage) with no worsening of NASH2 |
38 | % | 18 | % | ||||
| Resolution of NASH with no worsening of liver fibrosis3 |
24 | % | 9 | % | ||||
| Fibrosis Improvement and resolution of NASH4 |
22 | %* | 0 | % | ||||
| NAS reduction of >2 points with no worsening of liver fibrosis |
62 | %*** | 9 | % | ||||
*p=0.015; ***p<0.001
| 1 | Per protocol, analyzed using the “liver histologic population,” defined as the subset of enrolled patients who had valid, non-missing biopsy data at both baseline and week 24 (n=72). |
| 2 | Defined as patients having an improvement in liver fibrosis ³1 stage and having no worsening of hepatocellular ballooning, no worsening of lobular inflammation and no worsening of steatosis from baseline to week 24. |
| 3 | Defined as patients having a non-alcoholic fatty liver disease activity score, or NAS, of 0 or 1 for lobular inflammation and 0 for hepatocellular ballooning, with no worsening of fibrosis (no progression of NASH fibrosis stage) from baseline to week 24 (as defined by Clinical Research Network criteria). |
| 4 | Defined as patients having an improvement in liver fibrosis ³1 stage and having a NAS of 0 or 1 for lobular inflammation and 0 for hepatocellular ballooning at week 24. |
Efficacy data from a secondary analysis of patients with advanced liver fibrosis enrolled in Cohort 4 was also provided. In this patient population, 30% of patients with F3 liver fibrosis treated with 1 mg aldafermin achieved fibrosis improvement >1 stage without worsening of NASH compared to 0% in the placebo arm. A responder analysis conducted in patients with F3 liver fibrosis who achieved ³30% LFC reductions showed that 46% of patients treated with 1 mg aldafermin had fibrosis improvement of ³1 stage without worsening of NASH compared to 0% of placebo patients.
Similar to what has been observed in prior NASH clinical studies with aldafermin, Cohort 4 findings also demonstrated that patients treated with 1 mg aldafermin experienced a mean increase of 45 mg/dL LDL cholesterol, or LDL-C, at week two of treatment relative to baseline. Under the protocol, patients in both the placebo and aldafermin arms who experienced an increase in mean levels of LDL-C of at least 10 mg/dL at week two of treatment were directed to take rosuvastatin daily during the treatment period. LDL-C was effectively managed with the concomitant statin use and LDL-C levels for both the placebo and aldafermin arms were nearly identical at approximately 77 mg/dL at week 24. In addition, mean serum triglyceride levels were significantly reduced in patients treated with aldafermin versus placebo as early as week two and sustained through week 24.
S-3
Table of Contents
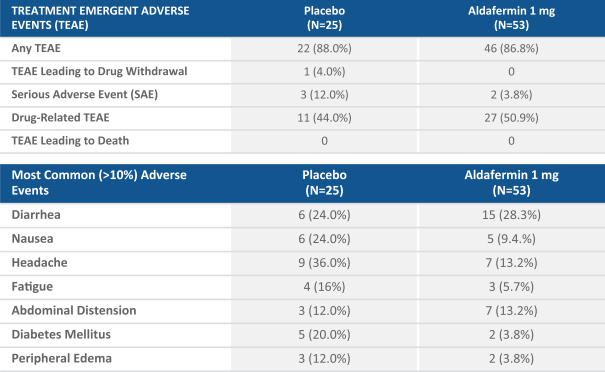
TREATMENT EMERGENT ADVERSE EVENTS (TEAE) Placebo (N=25) Aldafermin 1 mg (N=53) Any TEAE 22 (88.0%) 46 (86.8%) TEAE Leading to Drug Withdrawal 1 (4.0%) 0 Serious Adverse Event (SAE) 3 (12.0%) 2 (3.8%) Drug-Related TEAE 11 (44.0%) 27 (50.9%) TEAE Leading to Death 0 0 TREATMENT EMERGENT ADVERSE EVENTS (TEAE) Placebo (N=25) Aldafermin 1 mg (N=53) Any TEAE 22 (88.0%) 46 (86.8%) TEAE Leading to Drug Withdrawal 1 (4.0%) 0 Serious Adverse Event (SAE) 3 (12.0%) 2 (3.8%) Drug-Related TEAE 11 (44.0%) 27 (50.9%) TEAE Leading to Death 0 0
In Cohort 4, aldafermin had an overall adverse event profile that was similar to that of placebo, with no meaningful difference in gastrointestinal or pruritus adverse events in aldafermin compared to placebo. Serious adverse events, or SAEs, were also similar to placebo (aldafermin 4% versus 12% placebo), with all SAEs determined not to be related to treatment by the site investigator. Aldafermin was generally well tolerated and there were no study withdrawals due to adverse events, or AEs, in the aldafermin arm as compared to one withdrawal due to an adverse event in the placebo arm. Specific AE information is provided in the table below:
Finally, in November 2020, additional Cohort 4 data was presented showing that statistically significant reductions in C4, a marker of bile acid synthesis, and bile acids were observed with aldafermin compared to placebo. In patients with NASH, the rate-limiting enzyme for bile acid synthesis, CYP7A1, is upregulated and an associated increase in serum levels of bile acids is also observed.
MK-3655 in NASH
MK-3655 is an agonistic antibody selectively activating FGFR1c/KLB that we believe has the potential to be a once-monthly injectable insulin sensitizer for the treatment of NASH. In November 2018, Merck exercised its option for a license to further research, develop and commercialize MK-3655 and other FGFR1c/KLB agonists pursuant to our Collaboration Agreement. In Phase 1 clinical testing, MK-3655 demonstrated favorable tolerability and preliminary data has shown the agent is capable of reducing LFC and improving metabolic biomarkers in obese insulin resistant subjects with nonalcoholic fatty liver disease, or NAFLD, after a single dose. We conducted a Phase 1b randomized, open-label, parallel group trial to evaluate the safety, tolerability, PK and pharmacodynamics, or PD, of a single MK-3655 dose or the maximum approved dose of daily oral pioglitazone in 25 obese insulin resistant subjects with NAFLD. The Phase 1b clinical trial evaluated the ability of MK-3655 to decrease LFC to support the clinical development of MK-3655 in NASH. The primary objectives of the study were to
S-4
Table of Contents
evaluate changes from baseline in LFC as measured by magnetic resonance imaging-estimated proton density fat fraction at day 36 and changes from baseline in whole body insulin sensitivity at day 29 in subjects treated with MK-3655 as compared to pioglitazone. This study indicated that MK-3655 was well tolerated, with no SAEs and no AE leading to study discontinuation. All AEs observed during the course of the study were deemed mild, with increased appetite (12%) and injection site reaction (12%) being the only AEs reported in at least 10% of MK-3655-treated subjects.
In the fourth quarter of 2020, Merck initiated a Phase 2b trial of MK-3655 in patients with F2-F3 NASH.
NGM621 in Geographic Atrophy
NGM621 is an inhibitory antibody designed to bind and inactivate complement C3, a protein implicated in the pathology of geographic atrophy, or GA. We initiated a Phase 1 safety, tolerability and PK clinical trial of NGM621, administered via intravitreal, or IVT, injections, in patients with GA in the second half of 2019. NGM621 is currently being tested in the ongoing Phase 2 CATALINA trial in patients with GA to evaluate its effects on disease progression when given every four weeks or every eight weeks. The first patient was dosed in the CATALINA trial in July 2020. Merck has a one-time option to license NGM621 upon our completion of a proof-of-concept study in humans.
In November 2020, we announced results from our completed Phase 1 clinical study of NGM621. The primary objective of the Phase 1 trial was to assess the safety and tolerability of single and multiple IVT injections of NGM621 in patients with GA. Secondary objectives were to characterize the serum PK of single or multiple doses of NGM621. The study enrolled 15 patients across three single-ascending dose cohorts of NGM621, 2 mg, 7.5 mg and 15 mg, the maximum planned dose in the study, and a multiple dose cohort that received two 15 mg doses separated by four weeks. Patients were dosed sequentially and followed closely over 12 weeks. In the study, NGM621 was well tolerated, with no patients experiencing SAEs, drug-related AEs, intraocular inflammation, endophthalmitis or choroidal neovascularization. No dose-related safety patterns or concerns were reported. Ocular AEs observed were mild in severity and representative of those commonly associated with IVT injections. No vision-related safety signals were detected. On average, patients maintained their visual acuity over the 12-week follow-up study duration. The serum PK of NGM621 was linear and dose-proportional. Ocular PK/PD preclinical modeling conducted by Merck and us suggests that NGM621 may potentially achieve >90% reduction in free C3 in the eye for seven weeks following a single IVT dose of 15 mg. Taken together, we believe the PK profile of NGM621 demonstrated in the Phase 1 study and subsequent PK/PD preclinical modeling support the potential for up to an every eight week (or every other month) dosing regimen of NGM621 at the 15 mg dose level. NGM621 serum exposure was below concentrations expected to produce systemic complement inhibition after IVT injection of the 15 mg dose. No anti-drug antibodies were detected in any patient at any timepoint.
NGM120 in CACS and Cancer
NGM120 is an inhibitory antibody binding GFRAL that is designed to block the effects of elevated GDF15 levels on cancer anorexia/cachexia syndrome, or CACS, and potentially on tumors. NGM120 works by selectively inhibiting the interaction between GDF15 and its cognate receptor, GFRAL, through which the autonomic nervous system and, possibly, the neuroendocrine axis influence the body’s metabolism to propel the cachectic state, and potentially the tumors, in cancer patients that have high serum levels of GDF15. Merck has a one-time option to license NGM120 upon our completion of a proof-of-concept study in humans.
S-5
Table of Contents
In 2019, we completed a Phase 1 clinical trial in single (n=48) and multiple (n=44) ascending cohorts of NGM120 in healthy volunteers that assessed its safety, tolerability and PK profile. This clinical trial demonstrated that NGM120 was well tolerated at all doses studied and the pharmacokinetics supported once-monthly dosing. No SAEs or AEs were identified. The PK profile of NGM120 had a terminal half-life of approximately 35 days.
In the first quarter of 2020, we initiated a Phase 1a/1b clinical trial to assess the anti-CACS and anti-cancer effect of NGM120 in patients with advanced solid tumors. We recently completed enrollment in the Phase 1a dose finding portion of the study of NGM120 as a monotherapy in patients with select solid tumors and in the the Phase 1b dose finding portion of the study of NGM120 in combination with gemcitabine and abraxane in patients with metastatic pancreatic cancer. We expect to report data from the Phase 1a/1b dose finding study in the second half of 2021. A Phase 1b placebo-controlled expansion study of gemcitabine and abraxane and either NGM120 or placebo is planned in metastatic pancreatic carcinoma patients, and we anticipate initiating that study in the first quarter of 2021.
NGM707
NGM707 is a novel dual antagonist antibody that is designed to improve patient immune responses to tumors by inhibiting both the Immunoglobulin-like transcript 2, or ILT2 (also known as LILRB1), and Immunoglobulin-like transcript 4, or ILT4 (also known as LILRB2), receptors. NGM707 targets an epitope shared by both ILT2 and ILT4 to achieve inhibition of both receptor systems. We believe NGM707 has the potential to both reprogram ILT4-and ILT2-expressing myeloid cells to shift them from suppressing to activating immune detection and attacking tumors. ILT2 blockage may also reverse inhibition of certain ILT2-expressing lymphoid effector cells to promote further tumor response. Reversing myeloid suppression, or myeloid reprogramming, may represent a promising new therapeutic area of immuno-oncology aimed at increasing patient response rates to T cell checkpoint inhibitors.
ILT2/ILT4 receptors
ILT2 and ILT4 receptors expressed on myeloid cells are implicated in suppressing anti-tumor immune responses in the tumor microenvironment and may represent checkpoints that enable tumors to evade immune detection. Suppressive myeloid cells enriched with ILT2 and ILT4 receptors are upregulated in certain cancer types, while ILT2 is also expressed on natural killer, or NK, cells, B cells and a subset of highly cytolytic T cells. Of note, ILT2 and ILT4 are upregulated on macrophages in the tumor microenvironment of certain cancer patients that are non-responders to T cell checkpoint inhibitor therapy and, therefore, implicate them as T cell checkpoint inhibitor resistance mechanisms.
S-6
Table of Contents
Preclinical Studies and Planned Clinical Trials
Preclinical studies of NGM707 suggest that blockade of ILT4 reverses myeloid cell immune suppression, while blockade of ILT2 promotes NK and CD8+ T cell killing of tumor cells and activates macrophage phagocytosis of tumor cells. In addition, preclinical studies of NGM707 have shown that the dual blockade of ILT2 and ILT4 acts synergistically to reverse suppression of fragment crystallizable, or Fc, receptor signaling. In preclinical tests of monocytes from two individuals, the combination of NGM707 and pembrolizumab acted additively to increase T cell activation and cytokine secretion as shown in the mixed lymphocyte reactions’ graphics below.
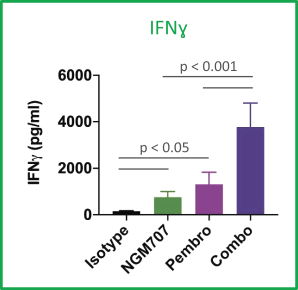

IFNg = Interferon Gamma; TNFa = Tumor Necrosis Factor alpha; Pembro= pembrolizumab; Combo = pembrolizumab and NGM707
All preclinical studies to enable an investigational new drug application, or IND, have been completed. We are preparing the final reports for a planned IND submission in early 2021, and plan to initiate a first-in-human study of NGM707 in mid-2021. Merck has a one-time option to license NGM707 upon our completion of a proof-of-concept study in humans.
NGM438
NGM438 is a novel antagonistic antibody that is designed to inhibit leukocyte-associated immunoglobulin-like receptor 1, or LAIR1, and promote immune detection and activation against advanced solid tumors. NGM438 has the potential to block the binding of all collagens tested with high potency, including tumor cell-derived collagens, which are the endogenous natural forms of collagen produced by the tumor that are believed to bind to LAIR1 to drive an immuno-suppressive environment surrounding the tumor. Reinvigoration of these collagen-suppressed immune cells may address a key resistance mechanism that limits responses to current immunotherapies.
LAIR1 receptor
LAIR1 is a collagen-binding inhibitory receptor expressed on immune cells that is implicated in immune suppression. LAIR1 and collagens are upregulated in multiple cancer types where collagens
S-7
Table of Contents
are produced by activated stromal cells. These stromal-derived suppressive factors are associated with poor responses to checkpoint inhibitors. For such tumors, formation of the LAIR1-collagen complex may act as a stromal checkpoint to both physically exclude immune cells from the tumor and impose signaling-based immune suppression. Inhibiting this stromal checkpoint represents a potentially promising new therapeutic strategy to treat cancer by promoting the remodeling of the tumor architecture that restricts T cell infiltration of the tumor cell mass and reversing immune suppression in the tumor microenvironment.
Preclinical Studies and Planned Clinical Trials
Preclinical studies suggest that NGM438 may have the potential to reprogram collagen-suppressed myeloid cells to a stimulatory phenotype, induce inflammatory cytokine production by myeloid and T cells and relieve collagen-based suppression of T cell proliferation. For example, in a preclinical model as shown in the graphics below, collagen suppressed the mixture of myeloid cells and T cells in mixed lymphocyte reactions while the administration of NGM438 in vitro reversed this T cell suppression in a dose-dependent manner.
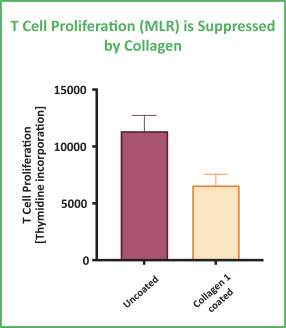
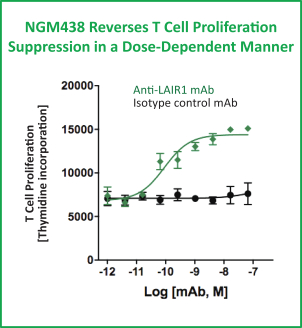
MLR = Mixed Lymphocyte Reaction; mAb = monoclonal antibodies
We plan to submit an IND in the second half of 2021 and expect to initiate a first-in-human study of NGM438 in the fourth quarter of 2021. Merck has a one-time option to license NGM438 upon our completion of a proof-of-concept study in humans.
Financial Update
Our consolidated financial statements for the year ended December 31, 2020 will not be available until after this offering is completed and consequently will not be available to you prior to investing in this offering. Based upon preliminary estimates and information available to us as of the date of this
S-8
Table of Contents
prospectus supplement, we expect to report that we had approximately $295 million of cash, cash equivalents and short-term marketable securities as of December 31, 2020. We have not yet completed our financial close process for the quarter and year ended December 31, 2020. This estimate of our cash, cash equivalents and short-term marketable securities as of December 31, 2020 is preliminary, has not been audited and is subject to change upon completion of our financial statement closing procedures and the audit of our consolidated financial statements. Additional information and disclosures would be required for a more complete understanding of our financial position and results of operations as of December 31, 2020.
Summary Risk Factors
Below is a summary of the material factors that make an investment in our common stock speculative or risky. Importantly, this summary does not address all of the risks that we face. Our business involves significant risks that may have a material adverse effect on our business, financial condition, results of operations, prospects and stock price. These risks are more fully described under “Risk Factors” beginning on page S-14 of this prospectus supplement and include, among others:
| • | our most advanced product candidate, aldafermin, is still in Phase 2 development and may fail to demonstrate safety and efficacy in ongoing and future clinical trials, may never achieve regulatory approval and may not be able to be successfully commercialized due to competition or other factors; |
| • | similarly, clinical trials of our other product candidates may fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or may otherwise fail to produce positive results; |
| • | we have incurred net losses every year since our inception, expect to incur significant and increasing operating losses and may never become profitable; |
| • | all of our revenue for recent periods has been received from a single collaboration partner, Merck, and Merck may choose not to extend the research phase of the collaboration beyond March 16, 2022 and, under certain circumstances, Merck may unilaterally terminate its funding of our research and development programs or shift the focus of its research and development funding; |
| • | we depend on our collaboration with Merck, and in the future may depend on collaborations with additional third parties, for the development and commercialization of our product candidates; |
| • | we will require additional capital following this offering to finance our operations, which may not be available to us on acceptable terms, or at all, and as a result, we may not complete the development and commercialization of our current product candidates or develop new product candidates; |
| • | we currently have no approved products or product revenue, and we will need to successfully complete preclinical and clinical testing of our product candidates before we can seek regulatory approval and potentially generate commercial sales; |
| • | our product candidates must undergo rigorous clinical trials before seeking regulatory approvals, which could delay or prevent commercialization of our product candidates; |
| • | the process of manufacturing aldafermin and our other biologic product candidates is complex, highly regulated and subject to several risks, including difficulties in production, particularly in scaling up and validating initial production and ensuring the avoidance of contamination; |
S-9
Table of Contents
| • | we rely on single source suppliers for the manufacture of our product candidates; |
| • | the regulatory approval processes of the U.S. Food and Drug Administration, or FDA, and comparable foreign regulatory authorities are lengthy, time-consuming and inherently unpredictable; |
| • | our future success depends in part on our ability to attract and retain highly skilled employees, including members of our current senior management team, especially Dr. Jin-Long Chen; |
| • | the COVID-19 pandemic continues to adversely impact our business and could materially and adversely affect our operations, as well as the businesses or operations of our manufacturers, clinical research organizations, or CROs, or other third parties with whom we conduct business; |
| • | we face substantial competition, which may result in others discovering, developing or commercializing products before, or more successfully than, us; |
| • | our success depends upon our ability to obtain and maintain intellectual property protection for our products and technologies, and we may not be able to protect our intellectual property rights throughout the world; |
| • | our principal stockholders, including entities affiliated with The Column Group, Merck and our management, own a substantial percentage of our stock and will be able to exert significant control over matters subject to stockholder approval; |
| • | we may not be able to obtain and maintain the relationships with third parties that are necessary to develop, commercialize and manufacture some or all of our product candidates; and |
| • | the market price of our common stock has been and may continue to be volatile, and you could lose all or part of your investment. |
Company Information
We were incorporated in Delaware in December 2007 and commenced operations in 2008. Our principal executive offices are located at 333 Oyster Point Blvd., South San Francisco, California 94080-7014, and our telephone number is (650) 243-5555. Our website address is http://www.ngmbio.com. The contents of and information accessible through our website are not incorporated into this prospectus supplement or the accompanying prospectus, and our reference to the URL for our website is intended to be an inactive textual reference only.
As used in this prospectus supplement and the accompanying prospectus, “NGM Biopharmaceuticals”, “NGM”, “we”, “us” and “our” refer to NGM Biopharmaceuticals, Inc. and its wholly-owned subsidiary taken as a whole. This prospectus supplement, the accompanying prospectus and the information incorporated herein by reference include trademarks, service marks and trade names owned by us or other companies. All trademarks, service marks and trade names included or incorporated by reference into this prospectus supplement, the accompanying prospectus or any related free writing prospectus are the property of their respective owners. We do not intend our use or display of other companies’ trademarks or trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies or products.
Implications of Being an Emerging Growth Company
We are an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012, as amended, or JOBS Act. We will remain an emerging growth company until the earliest to
S-10
Table of Contents
occur of: (i) the last day of the fiscal year in which we have more than $1.07 billion in annual revenue; (ii) the date we qualify as a “large accelerated filer,” under the rules of the SEC which, among other requirements, means that we have been subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, or Exchange Act, for at least twelve calendar months, we have filed at least one annual report under the Exchange Act, and the market value of our equity securities that is held by non-affiliates exceeds $700 million as of the prior June 30th; (iii) the issuance, in any three-year period, by us of more than $1.0 billion in non-convertible debt securities; and (iv) December 31, 2024.
As a result of this status, we may take advantage of certain exemptions from various public company reporting requirements, including not being required to have our internal control over financial reporting audited by our independent registered public accounting firm under Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and any golden parachute payments. In addition, the JOBS Act provides that an emerging growth company may take advantage of an extended transition period for complying with new or revised accounting standards, delaying the adoption of these accounting standards until they would apply to private companies unless we otherwise irrevocably elect not to avail itself of this exemption. However, we have chosen to irrevocably “opt out” of such extended transition period, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to not take advantage of the extended transition period for complying with new or revised accounting standards is irrevocable.
S-11
Table of Contents
THE OFFERING
| Common stock offered by us |
4,629,630 shares of our common stock. | |
| Common stock to be outstanding immediately after this offering |
73,564,397 shares, or 74,258,841 shares if the underwriters’ option to purchase additional shares is exercised in full. | |
| Underwriters’ option to purchase additional shares |
The underwriters have the option to purchase up to an additional 694,444 shares of our common stock, which they may exercise, in whole or in part, for a period of 30 days from the date of this prospectus supplement. | |
| Use of proceeds |
The net proceeds from the issuance of our common stock in this offering will be approximately $117.0 million or approximately $134.7 million if the underwriters exercise their option in full, after deducting underwriting discounts and commissions and estimated offering expenses payable by us. We intend to use the net proceeds from this offering for working capital and other general corporate purposes, which may include funding our pipeline of development programs, general and administrative activities and capital expenditures. See “Use of Proceeds” for additional information. | |
| Risk factors |
See “Risk Factors” beginning on page S-14 of this prospectus supplement for a discussion of factors that make an investment in our common stock speculative or risky. | |
| Nasdaq Global Select Market symbol |
Our common stock is listed on The Nasdaq Global Select Market under the symbol “NGM.” | |
Outstanding Shares
The number of shares of our common stock to be outstanding after this offering as stated above is based on 68,934,767 shares of our common stock outstanding as of September 30, 2020, and excludes as of that date:
| • | 10,651,475 shares of our common stock issuable upon the exercise of outstanding stock options with a weighted-average exercise price of $10.03 per share; |
| • | 6,309,573 shares of our common stock available for issuance or future grant under our Amended and Restated 2018 Equity Incentive Plan, or EIP; |
| • | 788,120 shares of our common stock available for issuance under our 2019 Employee Stock Purchase Plan, or ESPP; and |
| • | 21,930 shares of our common stock reserved for future issuance under our NGM Biopharmaceuticals Matching Plan, or the Matching Plan. |
The number of shares of common stock to be outstanding immediately following this offering does not reflect any automatic (or other) increases in the number of shares of common stock reserved for future issuance under the EIP, the ESPP or the Matching Plan, including the 2,823,565 shares of common stock automatically added to the EIP on January 1, 2021 in accordance with the terms of the EIP. In addition, the number of shares of common stock to be outstanding immediately following this
S-12
Table of Contents
offering as shown above also does not include up to $150.0 million of our common stock that remained available for sale at September 30, 2020 under our Open Market Sale AgreementSM, dated June 5, 2020, with Jefferies LLC, as agent, or the Sales Agreement. Between September 30, 2020 and the date of this prospectus supplement, 809,700 shares were sold under the Sales Agreement at an average price of $27.94 per share for net proceeds of $21.9 million, after deducting sales commissions. As of the date of this prospectus supplement, $127.4 million of common stock remained available to be sold under the Sales Agreement, subject to certain conditions as specified in the Sales Agreement.
Unless otherwise indicated, all information in this prospectus supplement assumes no exercise of the underwriters’ option to purchase additional shares of common stock from us.
S-13
Table of Contents
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below, together with all the other information included or incorporated by reference in this prospectus supplement and the accompanying prospectus and in any free writing prospectus that we may authorize for use in connection with this offering. The risks described below are not the only ones we face, but those that we consider to be material. There may be other unknown or unpredictable economic, business, competitive, regulatory or other factors that could have material adverse effects on our future results. Past financial performance may not be a reliable indicator of future performance, and historical trends should not be used to anticipate results or trends in future periods. If any of these risks actually occurs, our business, financial condition, results of operations or cash flow could be seriously harmed. This could cause the trading price of our common stock to decline, resulting in a loss of all or part of your investment. Please also read carefully the section below titled “Special Note Regarding Forward-Looking Statements.”
Risks Related to Our Financial Results and Capital Needs
We have incurred net losses every year since our inception, expect to incur significant and increasing operating losses and may never be profitable. Our stock is a highly speculative investment.
We are a biopharmaceutical company that was incorporated in 2007 and commenced operations in early 2008. Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that any potential product candidate will fail to demonstrate adequate effect and/or an acceptable safety profile, gain regulatory approval and become commercially viable. We have no products approved for commercial sale and have not generated any revenue from product sales to date, and we continue to incur significant research and development and other expenses related to our ongoing operations. As a result, we are not profitable and have incurred losses in each year since commencing operations. For the three and nine months ended September 30, 2020, our net losses were $29.8 million and $74.5 million, respectively, compared to net losses of $10.9 million and $26.9 million for the three and nine months ended September 30, 2019, respectively. As of September 30, 2020, we had an accumulated deficit of $270.6 million.
We have spent, and expect to continue to spend, significant resources to fund research and development of, and seek regulatory approvals for, our product candidates. We will require substantial additional capital to achieve our development and commercialization goals for our aldafermin program that is being conducted outside of the Merck collaboration, for any future programs that Merck does not opt to license under the Collaboration Agreement and that we choose to develop, for any Merck-licensed programs that we opt to co-develop and for any programs that Merck chooses to license under the Collaboration Agreement and later elects to terminate. If our research and development expenses for product candidates subject to the Merck collaboration exceed the funding caps provided in our Collaboration Agreement, which happened in the fiscal year ended December 31, 2020 and could potentially happen in the future, we will be required to devote our own financial resources toward the development of such product candidates or pause or suspend such development to remain within the funding caps. We expect to incur substantial and increasing operating losses over the next several years as our research, development, manufacturing, preclinical studies, clinical trial and related activities increase. As a result, our accumulated deficit will also increase significantly. We may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business, including those resulting from the evolving effects of the COVID-19 pandemic. The size of our future net losses will depend, in part, on the rate of future growth of our
S-14
Table of Contents
expenses and our ability to continue to generate revenue under the Merck collaboration and to generate revenue outside of the Merck collaboration. Our prior losses and expected future losses have had and will continue to have an adverse effect on our stockholders’ equity and working capital. Even if we eventually generate product revenue, we may never be profitable and, if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
All of our revenue for recent periods has been received from a single collaboration partner.
Since 2017, all of our revenue has been from our collaboration partner, Merck. Under the collaboration, Merck initially committed to reimburse us for research and development activities up to $50.0 million per year for at least five years. If our research and development expenses exceed $50.0 million in a given year and we are conducting IND-enabling or later-staged activities, Merck is required to elect either to reimburse up to an additional $25.0 million for use in funding IND-enabling or later-staged activities or to provide us with the equivalent value in in-kind services for preclinical and clinical development activities. In March 2019, Merck exercised its option to extend the initial five-year research and early development program, which we refer to as the research phase of the collaboration, for an additional two years through March 16, 2022. In connection with this extension, Merck agreed to continue to fund our research and development efforts during the extension at the same levels as existed during the five-year initial term and, in lieu of a $20.0 million extension fee that would have otherwise been payable to us, Merck agreed to make additional payments totaling up to $20.0 million in support of our research and development program activities under the collaboration across 2021 and the first quarter of 2022. Accordingly, the total Merck reimbursement for our research and development activities could reach up to $75.0 million per year through the current two-year extension of the research phase of the collaboration, plus up to $20.0 million in support of our research and development activities across 2021 and the first quarter of 2022. During the fiscal years ended December 31, 2019 and 2020, the total Merck reimbursement reached $75.0 million per year. Merck has the right to extend the research phase of the collaboration again through March 16, 2024. If our research and development expenses for product candidates subject to the Merck collaboration exceed the funding caps provided in our Collaboration Agreement, which happened in the fiscal year ended December 31, 2020 and could potentially happen in the future, we will be required to devote our own financial resources toward the development of such product candidates or pause or suspend such development to remain within the funding caps. For example, in 2020 we decided to suspend activities related to NGM386, NGM395 and NGM217 to concentrate our resources on our other Merck collaboration product candidates and aldafermin. In addition, if Merck elects not to exercise its remaining option to extend the research phase of the collaboration beyond March 16, 2022, which Merck may unilaterally elect to do at its sole discretion at any time before March 16, 2021, we would require significant additional capital in order to proceed with development and commercialization of any product candidates that had been subject to the Merck collaboration but Merck decides not to proceed with after termination of the research phase, or we will need to enter into additional collaboration or license agreements in order to fund such development and commercialization, which may not be possible, or we may be required to delay, scale back or discontinue development of such product candidates.
We currently have no source of product revenue and may never become profitable.
Our product candidates are in the early stages of development. To date, we have not generated any revenue from commercialization of our product candidates. We will not be able to generate product revenue unless and until one of our product candidates successfully completes clinical trials, receives regulatory approval and is successfully commercialized. As our product candidates are in Phase 2 trials or in earlier stages of development, we do not expect to receive revenue from those product candidates for a number of years, if ever. We may seek to obtain revenue from collaboration or licensing agreements with third parties. Other than our agreement with Merck, we currently have no such agreements that could provide us with material, ongoing future revenue and we may never enter into any such agreements.
S-15
Table of Contents
Our ability to generate future product revenue from our current or future product candidates also depends on a number of additional factors, including our or our current collaborator’s and potential future collaborators’ ability to:
| • | successfully complete research and clinical development of current and future product candidates; |
| • | establish and maintain supply and manufacturing relationships with third parties, and ensure adequate, scaled up and legally compliant manufacturing of bulk drug substances and drug products to maintain sufficient supply; |
| • | launch and commercialize any product candidates for which we obtain marketing approval, if any, and, if launched independently by us without a collaborator, successfully establish a sales force and marketing and distribution infrastructure; |
| • | demonstrate the necessary safety data (and, if accelerated approval is obtained, verify the clinical benefit) post-approval to ensure continued regulatory approval; |
| • | obtain coverage and adequate product reimbursement from third-party payors, including government payors, for any approved products; |
| • | achieve market acceptance for any approved products; |
| • | establish, maintain, protect and enforce our intellectual property rights; and |
| • | attract, hire and retain qualified personnel. |
In addition, because of the numerous risks and uncertainties associated with pharmaceutical product development, including that our product candidates may not advance through development or achieve applicable endpoints in clinical trials, we are unable to predict if or when we will achieve or maintain profitability. Even if we successfully complete development and regulatory processes, we anticipate incurring significant costs associated with launching and commercializing these products.
Even if we generate revenue from the sale of any of our products that may be approved, we may not become profitable and may need to obtain additional funding to continue operations. If we fail to become profitable or do not sustain profitability on a continuing basis, we may be unable to continue our operations at planned levels and be forced to reduce or cease our operations.
We will require additional capital following this offering to finance our operations, which may not be available to us on acceptable terms, or at all. As a result, we may not complete the development and commercialization of our current product candidates or develop new product candidates.
As a research and development company, our operations have consumed substantial amounts of cash since inception and we will require additional capital following this offering to finance our operations and pursue our strategy, which may not be available to us on acceptable terms, or at all. We expect our research and development expenses to increase substantially in connection with our ongoing activities, particularly to the extent that product candidates whose costs are not included in the Merck collaboration, such as aldafermin, advance in clinical development. In addition, we may be required to develop and implement additional clinical study policies and procedures to mitigate the evolving effects of the COVID-19 pandemic, which could significantly increase our research and development expenses. We believe that the net proceeds from this offering, together with our existing cash, cash equivalents and short-term marketable securities, will be sufficient to fund our operations for at least the next twelve months. We plan to continue to fund our operations and pursue our strategy through the Sales Agreement or other public or private equity or debt financings, government or other
S-16
Table of Contents
third-party funding, collaborations, strategic alliances, licensing arrangements or a combination of these. However, our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement that involves risks and uncertainties, and actual results could vary materially as a result of a number of factors, including the factors discussed elsewhere in this “Risk Factors” section.
We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect.
Our future funding requirements, both short- and long-term, will depend on many factors, including:
| • | the initiation, progress, timing, delays, costs and results of preclinical studies and clinical trials for our current product candidates and any future product candidates we may develop; |
| • | whether Merck exercises its option to license product candidates upon our completion of proof-of-concept studies for each such candidate in humans; |
| • | whether Merck terminates the research phase of the collaboration under pre-specified circumstances set forth in the Collaboration Agreement or terminates a program that it has licensed (such as Merck’s termination of its license for NGM395 and NGM386); |
| • | whether Merck exercises its remaining option to extend the research phase of the collaboration, which would trigger an extension payment to us; |
| • | whether we exceed the funding caps provided in our Collaboration Agreement, which happened in the fiscal year ended December 31, 2020 and could potentially happen in the future, which would require us to devote our own financial resources toward the development of programs and product candidates subject to the Merck collaboration during the current two-year extension of the research phase (and during a second extension should Merck unilaterally determine to trigger a second extension) or delay, scale back or discontinue such development to remain within the funding caps; |
| • | the outcome, timing and cost of seeking and obtaining regulatory approvals from the FDA and comparable foreign regulatory authorities, including the potential for such authorities to require that we perform more studies than those that we currently expect or to change their requirements on studies that had previously been agreed to; |
| • | the cost to establish, maintain, expand, enforce and defend the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with licensing, preparing, filing, prosecuting, defending and enforcing any patents or other intellectual property rights; |
| • | the effect of products that may compete with our product candidates or other market developments; |
| • | market acceptance of any approved product candidates, including product pricing and product reimbursement by third-party payors; |
| • | the cost of acquiring, licensing or investing in additional businesses, products, product candidates and technologies; |
| • | the cost and timing of selecting, auditing and potentially validating a manufacturing site for commercial-scale manufacturing; |
| • | the cost of establishing sales, marketing and distribution capabilities for our product candidates for which we may receive regulatory approval and that we determine to commercialize ourselves or in collaboration with partners; and |
S-17
Table of Contents
| • | the extent to which any of the foregoing costs are the responsibility of Merck. |
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our product candidates or intellectual property.
Unless and until we can generate a sufficient amount of revenue from approved products, we will require additional capital to discover, develop, obtain regulatory approval for and commercialize our current and future product candidates. We do not have any committed external source of funds, other than pursuant to our collaboration with Merck, which is limited in scope and duration, and may be unilaterally terminated by Merck under certain circumstances.
We plan to finance our future cash needs through the Sales Agreement or other public or private equity or debt offerings, government or other third-party funding, product collaborations, strategic alliances, licensing arrangements or a combination of these. Additional capital may not be available in sufficient amounts, on reasonable terms or when we need it, if at all, including pursuant to the Sales Agreement, and our ability to raise additional capital may be adversely impacted by worsening global economic conditions and the disruptions to, and volatility in, the credit and financial markets in the United States and worldwide resulting from, among other things, the evolving effects of the COVID-19 pandemic. Our existing stockholders could suffer dilution or be negatively affected by fixed payment obligations we may incur if we raise additional funds through the issuance of additional equity securities or debt, including equity securities already sold and that may in the future be sold pursuant to the Sales Agreement. Furthermore, these securities may have rights senior to those of our common stock and could contain covenants or protective rights that would restrict our operations and potentially impair our competitiveness, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business.
If we are unable to raise adequate additional capital, including pursuant to the Sales Agreement, we may not be able to expand our operations or otherwise capitalize on our business opportunities, our business and financial condition will be negatively impacted, and we may need to:
| • | significantly delay, scale back or discontinue research and discovery efforts and the development or commercialization of any product candidates, or cease operations altogether; |
| • | seek strategic alliances for research and development programs when we otherwise would not, at an earlier stage than we would otherwise desire or on terms less favorable than might otherwise be available; or |
| • | enter into product collaborations that would generally require us to relinquish, or license on potentially unfavorable terms, our rights to intellectual property, product candidates or products that we otherwise would seek to develop or commercialize ourselves, and we may not be able to enter into such agreements on acceptable terms, if at all. |
Accordingly, if we are unable to raise adequate additional capital, we may be prevented from pursuing development and commercialization efforts, which will have a material adverse effect on our business, operating results and prospects.
Our ability to use net operating loss carryforwards to offset taxable income could be limited.
We plan to use our current year operating losses to offset taxable income from any revenue generated from operations, including corporate collaborations. To the extent that our taxable income exceeds any current year operating losses, we plan to use our net operating loss carryforwards to offset income that would otherwise be taxable. Our net operating loss carryforwards generated in tax
S-18
Table of Contents
years ending on or prior to December 31, 2017 are only permitted to be carried forward for 20 years under applicable U.S. tax law. Under the Tax Cuts and Jobs Act, or the 2017 Tax Act, as modified by the Coronavirus Aid, Relief and Economic Security Act, or the CARES Act, our federal net operating losses generated in tax years beginning after December 31, 2017 may be carried forward indefinitely, but the deductibility of such federal net operating losses generated in tax years beginning after December 31, 2020, is limited to 80% of taxable income. It is uncertain if and to what extent various states will conform to the 2017 Tax Act or the CARES Act.
In addition, under Section 382 of the U.S. Internal Revenue Code of 1986, as amended, or the Code, and corresponding provisions of state law, if we experience an “ownership change,” generally defined as a greater than 50% change, by value, in equity ownership over a three-year period, our ability to use our pre-change net operating loss carryforwards to offset our post-change income may be limited. Due to our initial public offering, or the IPO, and subsequent shifts in our stock ownership, we have experienced ownership changes in the past and may experience ownership changes in the future as a result of this offering and other subsequent shifts in our stock ownership, some of which are outside our control. As a result, our use of federal net operating loss carryforwards could be limited. State net operating loss carryforwards may be similarly limited. In addition, at the state level, there may be periods during which the use of net operating losses is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. Any such limitations may result in greater tax liabilities than we would incur in the absence of such limitations and any increased liabilities could adversely affect our business, results of operations, financial position and cash flows.
Risks Related to Our Business and Industry
Our product candidates must undergo rigorous clinical trials before seeking regulatory approvals, which could delay or prevent commercialization of our product candidates.
All of our product candidates will be subject to rigorous and extensive clinical trials before we can seek regulatory approval from the FDA and comparable foreign regulatory authorities. Clinical trials may be delayed, suspended or terminated at any time for reasons including:
| • | ongoing discussions with the FDA or comparable foreign authorities regarding the scope or design of our clinical trials, including discussions with the FDA regarding initiation of our planned Phase 3 trial of aldafermin; |
| • | delays in obtaining, or the inability to obtain, required approvals from IRBs or other governing entities at clinical trial sites selected for participation in our clinical trials; |
| • | delays in enrolling participants into clinical trials, such as the slower pace of enrollment we have experienced, from time to time, in our ALPINE 4 and CATALINA trials, including as a result of the evolving effects of the COVID-19 pandemic; |
| • | delays in key trial activities and patient enrollment or diversion of healthcare resources as a result of the evolving effects of the COVID-19 pandemic, such as the slower pace of clinical trial site initiation in our ALPINE 4, CATALINA and NGM120 trials; |
| • | lower than anticipated retention rates of participants in clinical trials; |
| • | implementation of new, or changes to, guidance or interpretations from the FDA or comparable foreign authorities with respect to approval pathways for product candidates we are pursuing, such as draft guidance documents from the FDA for the development of products for the treatment of NASH that issued in 2018 and 2019 and from the European Medicines Agency, or EMA, that issued in 2018; |
| • | the need to repeat clinical trials as a result of inconclusive or negative results, poorly executed testing or changes in required endpoints by the FDA or comparable foreign authorities; |
S-19
Table of Contents
| • | insufficient supply or deficient quality of drug substance, drug product or other clinical trial material necessary to conduct our clinical trials; |
| • | unfavorable FDA or comparable foreign authority inspection and review of a clinical trial site or records of any clinical or preclinical investigation; |
| • | serious and unexpected drug-related adverse effects experienced by participants in our clinical trials; or |
| • | the placement of a clinical hold on a trial by the FDA or comparable foreign authorities. |
Positive or timely results from preclinical studies and early clinical trials do not ensure positive or timely results in late-stage clinical trials or product approval by the FDA or any other regulatory authority. Product candidates that show positive preclinical or early clinical results often fail in later stage clinical trials. Data obtained from preclinical and clinical activities is susceptible to varying interpretations, which could delay, limit or prevent regulatory approvals.
We have limited experience in conducting late-stage clinical trials required to obtain regulatory approval. We may not be able to conduct clinical trials at preferred sites, enlist clinical investigators, enroll sufficient numbers of participants or begin or successfully complete clinical trials in a timely fashion, if at all. Our current clinical trials may be insufficient to demonstrate that our potential products will be safe or effective. Additional clinical trials may be required if clinical trial results are negative or inconclusive, which will require us to incur additional costs and significant delays. If we do not successfully conduct clinical trials supporting the necessary regulatory approvals, we will not be able to generate product revenue and may not become profitable.
If clinical trials of our product candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or do not otherwise produce positive results, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we or our collaborators must conduct extensive preclinical studies and clinical trials to demonstrate the safety and efficacy of the product candidates in humans. Preclinical studies and clinical trials are expensive, take several years to complete and may not yield results that support further clinical development or product approvals. A failure of one or more clinical trials can occur at any stage of testing.
To date, the data supporting our drug discovery and development programs are derived from laboratory and preclinical studies and earlier-stage clinical trials. Despite the results reported in our Phase 1 and 2 clinical trials for aldafermin, in Phase 1 clinical trials for MK-3655 (NGM313), NGM621 and NGM120 and in preclinical studies for our other product candidates, including NGM707 and NGM438, future clinical trials in humans may show that one or more of our product candidates are not safe and effective, in which event we may need to abandon development of such product candidates. It is impossible to predict when or if any of our product candidates will prove safe and effective in humans or will receive regulatory approval. Owing in part to the complexity of biological pathways, these compounds might not demonstrate in patients the biochemical and pharmacological properties we anticipate based on laboratory studies or earlier-stage clinical trials, and they may interact with human biological systems or other drugs in unforeseen, ineffective or harmful ways.
Further, we expect that certain of our current product candidates will, and future product candidates may, require chronic administration. The need for chronic administration increases the risk that participants in our clinical trials will fail to comply with our dosing regimens. If participants fail to
S-20
Table of Contents
comply, we may not be able to generate clinical data in our trials acceptable to the FDA or foreign regulatory authorities. The need for chronic administration also increases the risk that our clinical drug development programs may not uncover all possible adverse events that patients who take our products may eventually experience. The number of patients exposed to treatment with, and the average exposure time to, our product candidates in clinical development programs may be inadequate to detect rare adverse events or chance findings that may only be detected once our products are administered to more patients and for longer periods of time.
If we are unable to successfully discover, develop or enable our collaborators to develop drugs that are effective and safe in humans, we will not have a viable business.
Success in preclinical studies or earlier-stage clinical trials may not be indicative of results in future clinical trials.
Success in preclinical studies and earlier-stage clinical trials does not ensure that later clinical trials will generate the same results or otherwise provide adequate data to demonstrate the effectiveness and safety of our product candidates. Similarly, preliminary data and interim results from clinical trials may not be predictive of final results. Frequently, product candidates that have shown promising results in early clinical trials have subsequently suffered significant setbacks in later clinical trials. For example, the results we obtained in our Phase 1 trials of aldafermin and in our completed Phase 2 trial, including the data from the fourth and final 24-week expansion cohort of that trial in patients with fibrosis stage F2 or F3 NASH, may not be indicative of the future results we obtain from our ongoing ALPINE 2/3 and ALPINE 4 trials and any Phase 3 trial.
Some of our clinical trials involve small patient populations, sometimes at single sites, and the results of these clinical trials may be subject to substantial variability and may not be indicative of either future interim results or final results. In addition, the design of a clinical trial can determine whether its results will support approval of a product, and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. Because we have limited experience designing clinical trials, we may be unable to design and execute a clinical trial to support regulatory approval.
In addition, there is a high failure rate for drugs and biologic products proceeding through clinical trials. In fact, many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in preclinical studies and earlier-stage clinical trials. Similarly, the outcome of preclinical studies may not predict the success of clinical trials. Moreover, data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent regulatory approval. In addition, we may experience regulatory delays or rejections as a result of many factors, including due to changes in regulatory policy during the period of our product candidate development. Any such delays could negatively impact our business, financial condition, results of operations and prospects.
If we continue to experience delays in clinical testing, we will be delayed in commercializing our product candidates, our costs may increase and our business may be harmed.
Conducting clinical trials for any of our product candidates for approval in the United States requires filing an IND application and reaching agreement with the FDA on clinical protocols, finding appropriate clinical trial sites and clinical investigators, securing approvals for such studies from the IRB for each such site, manufacturing clinical quantities of product candidates and supplying drug product to clinical trial sites. Currently, we have regulatory approval to conduct clinical trials in the United States for aldafermin for the treatment of NASH and primary biliary cholangitis, or PBC, for NGM621 for the treatment of GA secondary to AMD and for systemic administration and for NGM120
S-21
Table of Contents
for treatment of solid tumors and pancreatic cancer. We also have regulatory approval to conduct clinical trials in Australia for aldafermin for the treatment of NASH and for NGM395 for the treatment of metabolic syndrome. More recently, we obtained regulatory approval to conduct clinical trials in Germany, Spain, France, the United Kingdom, Belgium and Poland for aldafermin for the treatment of NASH.
We cannot guarantee that we will be able to successfully accomplish required regulatory activities or all of the other activities necessary to initiate and complete clinical trials. As a result, our preclinical studies and clinical trials may be extended, delayed or terminated, and we may be unable to obtain regulatory approvals or successfully commercialize our products. For example, we have experienced, from time to time, a slower pace of clinical trial site initiation and enrollment than originally anticipated in certain of our clinical trials, including the ALPINE 4, CATALINA and NGM120 trials, as a result of the evolving effects of the COVID-19 pandemic, and if the evolving effects of the COVID-19 pandemic become more severe, we could experience significant disruptions to our clinical development timelines, which would adversely affect our business, financial condition, results of operations and growth prospects. In any event, we do not know whether any of our clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. Our product development costs will increase if we continue to experience delays in clinical testing. Significant clinical trial delays could also shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do, which would impair our ability to successfully commercialize our product candidates and may harm our business, results of operations and prospects. Events that may result in a delay or failure to successfully complete clinical development include:
| • | delays in reaching agreement on acceptable terms with prospective CROs and clinical trial sites; |
| • | FDA comments on ongoing clinical trials and potential regulatory holds imposed if such comments are not adequately addressed; |
| • | deviations from the trial protocol by clinical trial sites and investigators, or failures to conduct the trial in accordance with regulatory requirements; |
| • | failure of third parties, such as CROs, to satisfy their contractual duties to us or meet expected deadlines; |
| • | delays in the testing, validation, manufacturing and delivery to clinical trial sites of the product candidates or other study materials; |
| • | delays in key trial activities, clinical trial site initiation and patient enrollment or diversion of healthcare resources as a result of the evolving effects of the COVID-19 pandemic; |
| • | for clinical trials in selected patient populations, delays in identification and auditing of central or other laboratories and the transfer and validation of assays or tests to be used to identify selected patients and test any patient samples; |
| • | delays in patient enrollment, such as the slower pace of enrollment we have experienced, from time to time, in our ALPINE 4 and CATALINA trials, including as a result of the evolving effects of the COVID-19 pandemic; |
| • | delays in patients completing a trial or returning for post-treatment follow-up; |
| • | delays caused by patients dropping out of a trial, including due to side effects, disease progression or concerns about the COVID-19 pandemic; |
| • | demonstration of a significant adverse safety or tolerability signal limiting the utility of the product candidate; |
S-22
Table of Contents
| • | changes in regulatory authority recommendations or guidance regarding development of drugs for a particular indication that we are pursuing, such as draft guidance documents from the FDA for the development of NASH that issued in 2018 and 2019 and from the EMA that issued in 2018; |
| • | withdrawal of clinical trial sites or investigators from our clinical trials for any reason, including as a result of changing standards of care or the ineligibility of a site to participate in our clinical trials; or |
| • | changes in government regulations or administrative actions or lack of adequate funding to continue the clinical trials. |
Our or our collaborators’ inability to timely complete clinical development could result in additional costs to us or impair our ability to generate product revenue or development, regulatory, commercialization and sales milestone payments and royalties on product sales.
If we or our partners are unable to enroll patients in clinical trials, we will be unable to complete these trials on a timely basis.
The timely completion of clinical trials largely depends on patient enrollment. Many factors affect patient enrollment, including:
| • | the size and nature of the patient population we enroll; |
| • | the number and location of clinical trial sites; |
| • | delays in enrollment due to travel or quarantine policies, behaviors or other factors related to COVID-19; |
| • | competition with other companies for clinical trial sites or patients; |
| • | the eligibility and exclusion criteria for the trial; |
| • | the design of the clinical trial; |
| • | inability to obtain and maintain patient consents; |
| • | risk that enrolled participants will drop out before completion; and |
| • | competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating. |
In particular, there is significant competition for recruiting NASH patients in clinical trials. In the first quarter of 2020, we announced that enrollment in our ALPINE 2/3 clinical trial of aldafermin had been delayed beyond our initial projections. In addition, clinical trial enrollment generally continues to be affected by the effects of the COVID-19 pandemic, including in our ongoing ALPINE 4 and CATALINA trials, including due to delays in additional clinical trial site initiation, suspension of enrollment at clinical trial sites or patient reluctance to participate in a clinical trial during quarantines or shelter-in-place orders or otherwise, particularly in medically vulnerable patient populations. We or our partners may be unable to enroll the patients we need to complete clinical trials on a timely basis, or at all.
We may not successfully identify, develop or commercialize our product candidates.
The success of our business depends primarily upon our ability to identify and validate new therapeutic candidates, and to identify, develop and commercialize protein and antibody therapeutics.
S-23
Table of Contents
Research programs to identify new product candidates require substantial technical, financial and human resources. Our research efforts may initially show promise in discovering potential new protein and antibody therapeutics, yet fail to yield product candidates for clinical development for a number of reasons, including:
| • | our research methodology may not successfully identify medically relevant protein or antibody therapeutics or potential product candidates; |
| • | our drug discovery efforts tend to identify and select novel, untested proteins in the particular disease indication we are pursuing, which we may fail to validate after further research work; |
| • | we may need to rely on third parties to generate protein or antibody candidates for some of our product candidate programs; |
| • | we may encounter product manufacturing difficulties that limit yield or produce undesirable characteristics that increase the cost of manufacturing our product candidates, cause delays or make the product candidates unmarketable; |
| • | our product candidates may cause adverse effects in patients or subjects, even after successful initial toxicology studies, which may make the product candidates unmarketable; |
| • | our product candidates may not demonstrate a meaningful benefit to patients or subjects; and |
| • | our partners may change their development profiles or plans for product candidates or abandon a therapeutic area, the development of a partnered product or the commercialization of any future approved partnered product. |
If any of these events occur, we may be forced to abandon our development efforts for one or more programs, which could have a material adverse effect on our business, operating results and prospects and could potentially cause us to cease operations. For example, we have suspended activities related to NGM386 and NGM217, and will be suspending activities related to NGM395 after the ongoing Phase 1 trial is completed, in all cases to concentrate our resources on our other product candidates. We may focus our efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful.
We are subject to many manufacturing risks, any of which could substantially increase our costs and limit supply of our product candidates and any future products.
To date, aldafermin and our other product candidates have been manufactured by third-party manufacturers solely for preclinical studies and relatively small clinical trials. These manufacturers may not be able to scale production to the larger quantities required for large clinical trials and for commercialization. The process of manufacturing aldafermin and our other product candidates is complex, highly regulated and subject to several risks, including:
| • | the process of manufacturing biologics is susceptible to product loss due to contamination, equipment failure, improper installation or operation of equipment, vendor or operator error and improper storage conditions. Even minor deviations from normal manufacturing processes could result in reduced production yields, product defects and other supply disruptions. If microbial, viral or other contaminations are discovered in our products or in the manufacturing facilities in which our products are made, the manufacturing facilities may need to be closed for an extended period of time to investigate and eliminate the contamination; |
| • | a third-party manufacturer of a product candidate subject to our collaboration with Merck may fail to qualify upon an audit by Merck; |
| • | the manufacturing facilities in which our products are made could be adversely affected by equipment failures, labor and raw material shortages, financial difficulties of our contract |
S-24
Table of Contents
| manufacturers, including as a result of the evolving effects of the COVID-19 pandemic, natural disasters, power failures, local political unrest and numerous other factors; and |
| • | any adverse developments affecting manufacturing operations or the scale up of manufacturing operations for our products may result in shipment delays, inventory shortages, lot failures, product withdrawals or recalls or other interruptions in the supply of our product candidates. We may also have to record inventory write-offs and incur other charges and expenses for product candidates or drug substances that fail to meet specifications, undertake costly remediation efforts or seek costlier manufacturing alternatives. |
We also have a single source of supply for most of our product candidates, including the drug substances used in manufacturing them. Single sourcing minimizes our leverage with our contract manufacturers, who may take advantage of our reliance on them to increase the pricing of their manufacturing services. Single sourcing also imposes a risk of interruption in supply in the event of manufacturing, quality or compliance difficulties. If one of our suppliers fails or refuses to supply us for any reason, it would take a significant amount of time and cost to implement and execute the necessary technology transfer to, and to qualify, a new supplier. The FDA or comparable foreign regulatory authority must approve manufacturers of drug substance and drug product. If there are any delays in qualifying new suppliers or facilities or a new supplier is unable to meet the requirements of the FDA or comparable foreign regulatory authority for approval, there could be a shortage of drug substance or drug product for use in clinical trials with respect to the affected product candidates.
In addition, the operations of our third-party manufacturers may be requisitioned, diverted or allocated by U.S. or foreign government orders such as under emergency, disaster and civil defense declarations in connection with the COVID-19 pandemic or otherwise. As an example, President Trump has invoked the Defense Production Act pursuant to which the U.S. government may, among other things, require domestic industries to provide essential goods and services needed for the national defense, such as drug material or other supplies needed to treat COVID-19 patients or to produce or distribute vaccines, which could require our third-party manufacturers to allocate manufacturing capacity in a way that delays or interrupts our supply of clinical trial material.
Specifically, we have entered into a Development and Manufacturing Services Agreement with Lonza Sales AG, or Lonza, for the production of Phase 3 and commercial supplies of the aldafermin drug substance. If Lonza or our drug product manufacturer are not able to provide us with sufficient quantities of aldafermin for our clinical trials on a timely basis, or at all, whether due to production shortages or other supply delays or interruptions resulting from the ongoing COVID-19 pandemic or otherwise, our clinical trials or regulatory approval may be delayed. In this regard, although significant portions of our research and development resources are focused, and will continue to be focused, on activities required to prepare aldafermin for potential regulatory approval for the treatment of NASH, including manufacturing of clinical trial material and preparation for potential Phase 3 testing, if Lonza and/or our drug product manufacturer experience difficulties in scaling production or experience product loss due to contamination, equipment failure, improper installation or operation of equipment, vendor or operator error or improper storage conditions, the potential Phase 3 testing of aldafermin would be delayed, perhaps substantially, which could materially and adversely affect our business. Moreover, our aldafermin drug product manufacturer has advised us that it could be required under orders of the U.S. government to allocate manufacturing capacity to the manufacture or distribution of COVID-19 vaccines. If Lonza Sales AG and/or our aldafermin drug product manufacturer become subject to acts or orders of U.S. or foreign government entities to allocate manufacturing capacity to the manufacture or distribution of COVID-19 vaccines or medical supplies needed to treat COVID-19 patients, this could also delay, perhaps substantially, the potential Phase 3 testing of aldafermin which could materially and adversely affect our business. Refer also to the risk factor entitled “Our business is currently adversely affected and could be materially and adversely affected in the future by the
S-25
Table of Contents
effects of disease outbreaks, epidemics and pandemics, including by the evolving effects of the COVID-19 pandemic. The COVID-19 pandemic continues to adversely impact our business and could materially and adversely affect our operations, as well as the businesses or operations of our manufacturers, CROs or other third parties with whom we conduct business.”
Each of our product candidates uses certain raw materials for its manufacture, such as reagents that support cell growth. Some of these materials only have a single supplier and are purchased as necessary without a long-term supply agreement in place. Any significant delay in the acquisition or decrease in the availability of these raw materials could considerably delay the manufacture of our product candidates, which could adversely impact the timing of any planned trials or the regulatory approvals of our product candidates.
The manufacture of biologic products is complex and requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of biologic products often encounter difficulties in production, particularly in scaling up and validating initial production and ensuring the avoidance of contamination. These problems include difficulties with production costs and yields, quality control, including stability of the product, quality assurance testing, operator error and shortages of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. We cannot ensure that any stability or other issues relating to the manufacture of our product candidates will not occur in the future.
We have limited process development capabilities and have access only to external manufacturing capabilities. We do not have, and we do not currently plan to acquire or develop, the facilities or capabilities to manufacture bulk drug substance or filled drug product for use in clinical trials or commercialization. Any delay or interruption in the supply of clinical trial material could delay the completion of clinical trials, increase the costs associated with maintaining clinical trial programs and, depending upon the period of delay, require us to commence new clinical trials at additional expense or terminate clinical trials completely.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval or limit the commercial profile of an approved label.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign regulatory authorities. Additional clinical trials may be required to evaluate the safety profile of our product candidates. Serious adverse events that were reported in the aldafermin treatment arms from our completed Phase 1 and Phase 2 clinical trials of aldafermin include: moderate dizziness, community acquired pneumonia, iron deficiency anemia, fractured finger, pneumonitis/alveolitis, acute pancreatitis, pneumonia, pleurisy, non-myocardial infarction cardiac arrest, chest pain, vertigo, headache, accelerated hypertension, kidney mass, bowel obstruction, bilirubin increase, cholangitis, progression of primary sclerosing cholangitis, or PSC, intervertebral discitis, rectal bleeding and post-biopsy bleeding. SAEs reported in our ongoing Phase 2 trials with aldafermin include gallbladder injury due to biopsy, suicide attempt and bronchogenic cyst, and no SAEs were deemed by investigators to be related to the treatment with aldafermin. In our completed Phase 1 and Phase 1b clinical trials of MK-3655 (NGM313), there were two reported SAEs in the MK-3655 (NGM313) treatment arms: cholecystitis and rectal bleeding due to hemorrhoids, both of which were deemed by the investigators to be unrelated to treatment with MK-3655 (NGM313). In our completed Phase 1 clinical trial of NGM120, there were two reported SAEs in the NGM120 treatment arms: renal colic and bipolar disorder, both of which were deemed by the investigators to be unrelated to treatment with NGM120. In our ongoing Phase 1a/1b trial of NGM120 evaluating NGM120 as a monotherapy in patients with
S-26
Table of Contents
select advanced tumors and in combination with two chemotherapeutic agents in patients with metastatic pancreatic cancer, there have been a number of SAEs, including sepsis, neutropenia, pulmonary embolism, pleural effusion, non-cardiac chest pain, renal failure, acute kidney injury and encephalopathy, none of which were deemed related to treatment with NGM120 after medical safety review. In our completed Phase 1 and ongoing Phase 2 clinical trials of NGM621, there have been no reported SAEs deemed related to treatment with NGM621.
Significant increases in serum levels of low-density lipoprotein, or LDL, cholesterol were observed in clinical trials of aldafermin in NASH and type 2 diabetes. The drug-induced changes in LDL cholesterol were brought back to baseline levels with concomitant statin use in NASH patients, however, sustained LDL cholesterol elevations in untreated patients can be associated with cardiovascular disease. We have not observed any significant changes in LDL cholesterol with aldafermin in trials we have conducted in patients with cholestatic liver disease, such as PBC and PSC.
Protein and antibody therapeutics can sometimes induce host immune responses that can cause the production of anti-drug-antibodies, or ADA. Our product candidates, including aldafermin, which is an engineered variant of the human FGF19 protein, are protein and antibody therapeutics. In some instances, certain ADA called neutralizing antibodies can neutralize the therapeutic effects of the treatment. ADA can also sometimes cross react with substances naturally occurring in a subject’s body (in the case of aldafermin, FGF19), which can cause unintended effects, including potential impacts on efficacy and, in rare cases, even adverse events. One subject in a Phase 2 clinical trial investigating aldafermin as an intervention for type 2 diabetes developed ADA against aldafermin; however, this patient did not demonstrate any biochemical or clinically relevant safety concerns while in the study, and we have not identified any safety concerns while monitoring the subject following the study. In the Phase 2 PBC extension clinical trial of aldafermin, 18 of the 36 subjects tested positive for aldafermin-specific ADA at one or more time points using a preliminary assay. Six of these 36 subjects tested positive for neutralizing antibodies using an assay that was subsequently validated, of which two subjects developed antibodies that appeared to cross-react with their naturally occurring FGF19. These subjects have not demonstrated any biochemical or clinically relevant safety signals that were different from observations in subjects that did not generate ADA against aldafermin. We are developing an assay to measure the presence of ADA against aldafermin for our ongoing NASH program, which will need to be evaluated by regulatory agencies. The subjects who were found to develop ADA in both the type 2 diabetic and PBC populations may not be predictive of future test results in NASH patients due to differences in disease setting, study design, dose regimen and the use of a different ADA assay. If we are required to do substantial additional testing as a result of the detection of high amounts of ADA in subjects using aldafermin or any other product candidate, the costs of our clinical trials may increase.
Future results of our trials could reveal a high and unacceptable severity and prevalence of side effects or ADAs that have negative effects or other unexpected characteristics. In such an event, we may need to suspend or terminate our trials or the FDA or comparable foreign regulatory authorities could order us to cease clinical trials or deny approval of our product candidates for any or all targeted indications. Drug-related side effects could affect patient recruitment, the ability of enrolled subjects to complete the trial or result in potential product liability claims. Any of these occurrences could materially and adversely affect our business, financial condition and prospects.
Our most advanced clinical-stage product candidate, aldafermin, is a modified version of a human hormone that has been associated with liver cancer in rodent testing.
Aldafermin is an engineered variant of the human hormone FGF19 that has been associated with liver cancer in rodent testing. The IND that we filed in February 2014 for type 2 diabetes was placed on
S-27
Table of Contents
clinical hold by the FDA Division of Metabolism and Endocrinology Products pending receipt of additional information relating to the potential risk of proliferative effects of aldafermin in the livers of non-human primates and mice based on concerns relating to the observation that human FGF19 can induce hepatocellular proliferation in rodents. We withdrew this IND in January 2015, as we determined that we would not further study aldafermin in type 2 diabetes after we analyzed the results of the Phase 2 clinical trial of aldafermin in type 2 diabetes and made the determination to pursue NASH and other liver indications. To date, the FDA Division of Hepatology and Nutrition, which is responsible for the NASH indication, has not requested any additional information regarding the potential for aldafermin to induce hepatocellular proliferation. We have received feedback from the FDA Carcinogenicity Assessment Committee that our preclinical data through six-month chronic toxicology studies in mice and monkeys support a single species, two-year carcinogenicity assessment in rats. The human hormone and the rodent ortholog for FGF19 share a sequence identity of approximately 50%, which means that the results of these studies of aldafermin in rats are not necessarily predictive of the potential risk of carcinogenicity in humans. To our knowledge, neither FGF19 nor any variant thereof other than aldafermin has ever been tested in humans. We believe we have identified a modified version of FGF19 that does not exhibit the cancer-causing effects of native human FGF19 in rodents. We believe that aldafermin will have a superior therapeutic profile to FGF19 based on preclinical data showing reduced fasting blood glucose levels, fed insulin levels and bile acid suppression in animals. However, we may be incorrect in these beliefs, and we cannot be sure that regulators will view our product candidate as safe or that physicians will view our product candidates as superior to alternative treatments. Concerns about the association between FGF19 and liver cancer could have an adverse effect on our ability to develop and commercialize aldafermin.
We have no experience in sales, marketing and distribution and may have to enter into agreements with third parties to perform these functions, which could prevent us from successfully commercializing our product candidates.
We currently have no sales, marketing or distribution capabilities. To commercialize our product candidates outside of the Merck collaboration, or to commercialize products subject to the Merck collaboration for which we may in the future exercise our co-detailing rights in the United States, we must either develop our own sales, marketing and distribution capabilities, which will be expensive and time-consuming, or make arrangements with third parties to perform these services for us. If we exercise our co-detailing rights in the United States with respect to the Merck collaboration, we will be responsible for the costs of fielding such a sales force, subject to partial offset pursuant to the formula by which profits are allocated, and the risks of attracting, retaining, motivating and ensuring the compliance of such a sales force with the various requirements of the Merck collaboration and applicable law. If we decide to market any of our products on our own, we will have to commit significant resources to developing a marketing and sales force and supporting distribution capabilities. If we decide to enter into arrangements with third parties for performance of these services, we may find that they are not available on terms acceptable to us, or at all. If we are not able to establish and maintain successful arrangements with third parties or build our own sales and marketing infrastructure, we may not be able to commercialize our product candidates, which would adversely affect our business and financial condition.
We may fail to select or capitalize on the most scientifically, clinically and commercially promising or profitable product candidates.
We have limited technical, managerial and financial resources to determine which of our product candidates should proceed to initial clinical trials, later-stage clinical development and potential commercialization. We may make incorrect determinations in allocating resources among these product candidates. Our decisions to allocate our research and development, management and financial resources toward particular product candidates or therapeutic areas may not lead to the
S-28
Table of Contents
development of viable commercial products and may divert resources from better opportunities. Similarly, our decisions to delay or terminate drug development programs, such as our decision to suspend activities related to NGM386, NGM395, NGM621 administered systemically and NGM217 to concentrate our resources on our other product candidates, may also be incorrect and could cause us to miss valuable opportunities.
Under our Collaboration Agreement with Merck, we have the right, exercisable during a specified period prior to the commencement of Phase 3 clinical testing of the applicable product candidate, to convert our economic participation from a milestones and net sales royalty arrangement into a cost and profit share arrangement. If we exercise the cost and profit share right, we have the ability to participate in a co-detailing relationship in the United States. Due to the limited exercise period, we may have to choose whether a product candidate will be subject to a cost and profit share arrangement before we have as much information as we would like, including whether and when such program may receive FDA approval of the applicable biologics license application, or BLA. As a result of such incomplete information or due to incorrect analysis by us, we may select a cost and profit share program that later proves to have less commercial potential than an alternative, or none at all, or may pass on a cost and profit sharing program that proves commercially successful.
We must attract and retain highly skilled employees in order to succeed. If we are not able to retain our current senior management team, especially Dr. Jin-Long Chen, or continue to attract and retain qualified scientific, technical and business personnel, our business will suffer.
To succeed, we must recruit, retain, manage and motivate qualified clinical, scientific, technical and management personnel and we face significant competition for experienced personnel. If we do not succeed in attracting and retaining qualified personnel, particularly at the management level, it could adversely affect our ability to execute our business plan and harm our operating results. An important element of our strategy is to take advantage of the research and development expertise of our current management. The loss of any one of our executive officers, including, in particular, Dr. Jin-Long Chen, our Chief Scientific Officer, could result in a significant loss in the knowledge and experience that we, as an organization, possess and could cause significant delays, or outright failure, in the development and further commercialization of our product candidates.
To fully realize the research and development support committed under our collaboration with Merck, we will need to maintain a significant number of qualified research and development personnel. There is intense competition for qualified personnel, including management, in the technical fields in which we operate, and we may not be able to attract and retain qualified personnel necessary for the successful research, development and future commercialization, if any, of our product candidates. In particular, the hiring environment in the San Francisco Bay Area, where we are headquartered, is extremely competitive. Many of the other pharmaceutical companies that we compete against for qualified personnel have greater financial and other resources, different risk profiles and a longer history in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high-quality candidates than what we have to offer. If we are unable to continue to attract and retain high-quality personnel, the rate and success with which we can discover and develop product candidates and our business will be limited.
Disputes under key agreements or conflicts of interest with our scientific advisors or clinical investigators could delay or prevent development or commercialization of our product candidates.
Any agreements we have or may enter into with third parties, such as collaboration, license, supplier, manufacturing, sponsored research, CRO or clinical trial agreements, may give rise to
S-29
Table of Contents
disputes regarding the rights and obligations of the parties. Disagreements could develop over contract interpretation, rights to ownership or use of intellectual property, the scope and direction of research and development, the approach for regulatory approvals or commercialization strategy. We are conducting research programs in a range of therapeutic areas, and our pursuit of these opportunities could result in conflicts with the other parties to these agreements that may be developing or selling pharmaceuticals or conducting other activities in these same therapeutic areas. Any disputes or commercial conflicts could lead to the termination of our agreements, delay progress of our product development programs, compromise our ability to renew agreements or obtain future agreements, lead to the loss of intellectual property rights, result in increased financial obligations for us or result in costly litigation.
We work with outside scientific advisors and collaborators at academic and other institutions that assist us in our research and development efforts. Our scientific advisors are not our employees and may have other commitments that limit their availability to us. If a conflict of interest between their work for us and their work for another entity arises, we may lose their services.
We may encounter difficulties in managing our growth, which could adversely affect our operations.
Since executing the Collaboration Agreement in 2015, we have significantly increased our headcount and advanced our pipeline and the complexity of our operations, which has placed a strain on our administrative and operational infrastructure. We expect this strain to continue as we seek to maintain our growth and seek to obtain and manage relationships with third parties. Our ability to manage our operations and growth effectively depends upon the continual improvement of our procedures, remote work policies, reporting systems and operational, financial and management controls, particularly in light of the evolving effects of the COVID-19 pandemic. We may not be able to expand or identify sufficiently-sized facilities to accommodate our growth, particularly given our location in South San Francisco, California and the current high demand for, and restricted supply of, research and development facilities in this market. We may not be able to implement administrative and operational improvements in an efficient or timely manner and may discover deficiencies in existing systems and controls. If we do not meet these challenges, we may be unable to take advantage of market opportunities, execute our business strategies or respond to competitive pressures, which in turn may slow our growth or give rise to inefficiencies that would increase our losses.
We may acquire additional assets, intellectual property and complementary businesses in the future. Acquisitions involve many risks, any of which could materially harm our business, including the diversion of management’s attention from core business concerns, failure to effectively exploit acquired assets or intellectual property, failure to successfully integrate the acquired business or realize expected synergies or the loss of key employees from either our business or the acquired businesses.
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than us.
The biopharmaceutical industry is intensely competitive and subject to rapid and significant technological change. Our competitors include multinational pharmaceutical companies, specialized biotechnology companies and universities and other research institutions. A number of pharmaceutical companies, including AbbVie, Allergan, AstraZeneca, Bayer, Boerhinger Ingelheim, Bristol-Myers Squibb, Eisai, Eli Lilly, GlaxoSmithKline, Johnson & Johnson, Merck, Novartis, Novo Nordisk, Pfizer, Roche, Sanofi and Takeda, as well as large and small biotechnology companies such as 89Bio, Akero, Albireo, Alentis, Amgen Inc., or Amgen, Apellis, Ascletis, Axcella, Bird Rock, Can-Fite, Cirius, Enanta, Galectin, Galmed, Gilead, Glympse, Immuron, Intercept, Inventiva, Iveric, Madrigal, MannKind,
S-30
Table of Contents
MediciNova, Metacrine, Mirum, North Sea, Promethera, Salix, Scholar Rock, Seal Rock, Terns, Tiziana, Viking and Vivus, are pursuing the development or marketing of pharmaceuticals that target the same diseases that are targeted by our most advanced product candidates. It is probable that the number of companies seeking to develop products and therapies for the treatment of liver and metabolic diseases, retinal diseases and cancer will increase. Many of our competitors have substantially greater financial, technical, human and other resources than we do and may be better equipped to develop, manufacture and market technologically superior products. In addition, many of these competitors have significantly greater experience than we have in undertaking preclinical studies and human clinical trials of new pharmaceutical products and in obtaining regulatory approvals of human therapeutic products. Accordingly, our competitors may succeed in obtaining FDA approval for superior products or for other products that would compete with our product candidates. Many of our competitors have established distribution channels and commercial infrastructure to support the commercialization of their products, whereas we have no such channel or capabilities. In addition, many competitors have greater name recognition and more extensive collaborative relationships. Smaller and earlier-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies.
Our competitors may obtain regulatory approval of their products more rapidly than us or may obtain patent protection or other intellectual property rights that limit our ability to develop or commercialize our product candidates. Our competitors may also develop drugs that are more effective, more convenient, more widely used and less costly or have a better safety profile than our products and these competitors may also be more successful than us in manufacturing and marketing their products. If we are unable to compete effectively against these companies, then we may not be able to commercialize our product candidates or achieve a competitive position in the market. This would adversely affect our ability to generate revenue. Our competitors also compete with us in recruiting and retaining qualified scientific, management and commercial personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
There are no currently approved therapies for NASH. Although we believe there are no approved therapies that specifically target the signaling pathways that our current product candidates are designed to modulate or inhibit, there are numerous currently approved therapies for treating the same diseases or indications, other than NASH, for which our product candidates may be useful and many of these currently approved therapies act through mechanisms similar to our product candidates. Many of these approved drugs are well-established therapies or products and are widely accepted by physicians, patients and third-party payors. Some of these drugs are branded and subject to patent protection, and others are available on a generic basis. Insurers and other third-party payors may also encourage the use of generic products or specific branded products. We expect that if our product candidates are approved, they will be priced at a significant premium over competitive generic products, including branded generic products. This may make it difficult for us to differentiate our products from currently approved therapies, which may adversely impact our business strategy. In addition, many companies are developing new therapeutics, and we cannot predict what the standard of care will be as our product candidates progress through clinical development.
If aldafermin or MK-3655 (NGM313) were approved for the treatment of NASH, future competition could also arise from products currently in development, including: cenicriviroc, an immunomodulator that blocks CCR2 and CCR5, from Allergan; firsocostat, an ACC inhibitor, and cilofexor, an FXR agonist, both from Gilead; OCA, an FXR agonist, from Intercept; resmetirom, a beta-thyroid hormone receptor agonist, from Madrigal; pegbelfermin, a PEGylated FGF21 analog, from Bristol-Myers Squibb; AKR-001, an Fc conjugated FGF21 analog, from Akero; FXR agonists from Metacrine; FXR agonists from Novartis; a beta-thyroid hormone receptor agonist from Viking; semaglutide, a GLP-1 analog, from Novo Nordisk; and lanifibranor, a pan-PPAR agonist from Inventiva. The foregoing competitive
S-31
Table of Contents
risks apply to aldafermin, any variants of aldafermin, including the second-generation, half-life extended version of FGF19 we are currently developing, and MK-3655 (NGM313).
Our product candidates may not achieve adequate market acceptance among physicians, patients, healthcare payors and others in the medical community necessary for commercial success.
Even if our product candidates receive regulatory approval, they may not gain adequate market acceptance among physicians, patients, healthcare payors and others in the medical community. Demonstrating the safety and efficacy of our product candidates and obtaining regulatory approvals will not guarantee future revenue. The degree of market acceptance of any of our approved product candidates will depend on a number of factors, including:
| • | the efficacy and safety profile of the product candidate as demonstrated in clinical trials; |
| • | the timing of market introduction of the product candidate, as well as competitive products; |
| • | the clinical indications for which the product candidate is approved; |
| • | acceptance of the product candidate as a safe and effective treatment by clinics and patients; |
| • | the potential and perceived advantages of the product candidate over alternative treatments, including any similar generic treatments; |
| • | the cost of treatment relative to alternative treatments; |
| • | the availability of coverage and adequate reimbursement and pricing by third parties and government authorities as described below; |
| • | the relative convenience and ease of administration; |
| • | the frequency and severity of adverse events; |
| • | the effectiveness of sales and marketing efforts; and |
| • | any unfavorable publicity relating to the product candidate. |
Sales of medical products also depend on the willingness of physicians to prescribe the treatment, which is likely to be based on a determination that the products are safe, therapeutically effective and cost effective. In addition, the inclusion or exclusion of products from treatment guidelines established by various physician groups and the viewpoints of influential physicians can affect the willingness of other physicians to prescribe the treatment. We cannot predict whether physicians, physicians’ organizations, hospitals, other healthcare providers, government agencies or private insurers will determine that our products are safe, therapeutically effective and cost effective as compared with competing treatments. If any product candidate is approved but does not achieve an adequate level of acceptance by such parties, we may not generate or derive sufficient revenue from that product candidate and may not become or remain profitable.
Even if we commercialize any of our product candidates, alone or with our partners, these products may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, which could harm our business.
The regulations that govern marketing approvals, pricing and reimbursement for new drug products vary widely from country to country. Current and future legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. In many regions, including Europe, Japan and Canada, the pricing of prescription drugs is
S-32
Table of Contents
controlled by the government. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. Regulatory agencies in those countries could determine that the pricing for our products should be based on prices of other commercially available drugs for the same disease, rather than allowing us to market our products at a premium as new drugs. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay or limit our commercial launch of the product, possibly for lengthy time periods, which could negatively impact the revenue we generate from the sale of the product in that particular country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if our product candidates obtain marketing approval.
Our commercial success also depends on coverage and adequate reimbursement of our product candidates by third-party payors, including government payors, private health insurers, health maintenance organizations and other organizations, which may be difficult or time-consuming to obtain, may be limited in scope and may not be obtained in all jurisdictions in which we may seek to market our products. Governments and private insurers closely examine medical products to determine whether they should be covered by reimbursement and, if so, the level of reimbursement that will apply. Government authorities and other third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. We cannot be sure that coverage and reimbursement will be available for any product that we or our partners commercialize and, if reimbursement is available, what the level of reimbursement will be. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which we or our partners obtain marketing approval. If coverage and reimbursement are not available or reimbursement is available only to limited levels, we and our partners may not be able to successfully commercialize any product candidate for which marketing approval is obtained.
There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that a drug will be paid for in all cases or at a rate that covers our costs, including costs of research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may only be temporary. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
The advancement of healthcare reform may negatively impact our ability to profitably sell our product candidates, if approved.
Third-party payors, whether domestic or foreign, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. The United States and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare
S-33
Table of Contents
system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product for which we obtain marketing approval.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or, collectively, the ACA, was enacted, which includes measures that have significantly changed the way health care is financed by both governmental and private insurers. There remain executive, judicial and congressional challenges to certain aspects of the ACA.
In addition, while Congress has not passed comprehensive legislation repealing the ACA, it has introduced legislation to modify certain provisions. Congress will likely consider other legislation to modify or replace additional elements of the ACA. It is unclear how these efforts to repeal and replace the ACA, or other appeals, will impact the ACA and our business. For example, the 2017 Tax Act repealed the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage that is commonly referred to as the “individual mandate.” In December 2019, a U.S. District Court upheld a ruling that the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. The Supreme Court of the United States is currently reviewing this case, although it is unclear when a decision will be made. It is also unclear how such litigation and other efforts to challenge, repeal, or replace the ACA will impact the ACA or our business.
Other legislative changes that have affected or may affect our industry include the Budget Control Act of 2011 which has triggered automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of, on average, 2% per fiscal year through 2030 unless Congress takes additional action.
Recently, there has also been increasing executive, legislative and enforcement interest in the United States with respect to drug pricing practices. There have been several recent U.S. congressional inquiries, presidential executive orders, and proposed and enacted legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs and reform government program reimbursement methodologies for drugs. We expect that the healthcare reform measures that have been adopted and may be adopted in the future may result in more rigorous coverage criteria and additional downward pressure on the price that we receive for any approved product and could seriously harm our future revenues. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our products.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. It is also possible that additional governmental action is taken in response to the COVID-19 pandemic. Such reforms could have an adverse effect on anticipated revenue from product candidates that we may successfully develop and for which we may obtain regulatory approval and may affect our overall financial condition and ability to develop product candidates.
We cannot predict the likelihood, nature or extent of healthcare reform initiatives that may arise from future legislation or administrative action, particularly as a result of the recent presidential election. If we or any third parties we may engage are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or such third parties are not able to maintain regulatory compliance, our product candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability.
S-34
Table of Contents
Our international operations may expose us to business, regulatory, political, operational, financial, pricing and reimbursement risks associated with doing business outside of the United States.
Our business is subject to risks associated with conducting business internationally. Some of our suppliers and clinical trial sites are located outside of the United States. Furthermore, if we, Merck or any future collaborator succeeds in developing any of our product candidates, we intend to market them in the European Union, or the EU, and other jurisdictions in addition to the United States. If approved, we, Merck or any future collaborator may hire sales representatives and conduct physician and patient association outreach activities outside of the United States. Doing business internationally involves a number of challenges and risks, including but not limited to:
| • | multiple, conflicting and changing laws and regulations, such as privacy regulations, tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses; |
| • | failure by us to obtain and maintain regulatory approvals for the use of our products in various countries; |
| • | rejection or qualification of foreign clinical trial data by the competent authorities of other countries; |
| • | delays or interruptions in the supply of clinical trial material resulting from any events affecting raw material supply or manufacturing capabilities abroad, including those that may result from the ongoing COVID-19 pandemic; |
| • | additional potentially relevant third-party patent rights; |
| • | complexities and difficulties in obtaining, maintaining, protecting and enforcing our intellectual property; |
| • | difficulties in staffing and managing foreign operations; |
| • | complexities associated with managing multiple payor reimbursement regimes, government payors or patient self-pay systems; |
| • | limits on our ability to penetrate international markets; |
| • | financial risks, such as longer payment cycles, difficulty collecting accounts receivable, the impact of local and regional financial crises on demand and payment for our products and exposure to foreign currency exchange rate fluctuations; |
| • | natural disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, including COVID-19 and related shelter-in-place orders, travel, social distancing and quarantine policies, boycotts, curtailment of trade and other business restrictions; |
| • | regulatory and compliance risks that relate to anti-corruption compliance and record-keeping that may fall within the purview of the U.S. Foreign Corrupt Practices Act, its accounting provisions or its anti-bribery provisions or provisions of anti-corruption or anti-bribery laws in other countries. |
Any of these factors could harm our ongoing international clinical operations and supply chain, as well as any future international expansion and operations and, consequently, our business, financial condition, prospects and results of operations.
S-35
Table of Contents
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and will face an even greater risk if we or our collaborator commercializes any resulting products. Product liability claims may be brought against us by subjects enrolled in our clinical trials, patients, healthcare providers or others using, administering or selling our products. If we cannot successfully defend ourselves against claims that our product candidates or products that we may develop caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| • | decreased demand for any product candidates or products that we may develop; |
| • | termination of clinical trial sites or entire trial programs; |
| • | injury to our reputation and significant negative media attention; |
| • | withdrawal of clinical trial participants; |
| • | significant costs to defend the related litigation; |
| • | substantial monetary awards to trial subjects or patients; |
| • | loss of revenue; |
| • | diversion of management and scientific resources from our business operations; and |
| • | the inability to commercialize any products that we may develop. |
Our clinical trial liability insurance coverage may not adequately cover all liabilities that we may incur. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise. Our inability to obtain product liability insurance at an acceptable cost or to otherwise protect against potential product liability claims could prevent or delay the commercialization of any products or product candidates that we develop. We intend to expand our insurance coverage for products to include the sale of commercial products if we obtain marketing approval for our product candidates in development, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. If we are sued for any injury caused by our products, product candidates or processes, our liability could exceed our product liability insurance coverage and our total assets. Claims against us, regardless of their merit or potential outcome, may also generate negative publicity or hurt our ability to obtain physician endorsement of our products or expand our business.
Our relationships with healthcare providers, customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse, transparency and other healthcare laws and regulations, which, if violated, could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm, administrative burdens and diminished profits and future earnings.
Healthcare providers, including physicians, and third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we or our collaborator obtains marketing approval. Our arrangements with healthcare providers, third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we research,
S-36
Table of Contents
market, sell and distribute our products for which we or our collaborator obtains marketing approval. Restrictions under applicable federal and state healthcare laws and regulations, include the following:
| • | the federal Anti-Kickback Statute prohibits persons from, among other things, knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, the referral of an individual for the furnishing or arranging for the furnishing, or the purchase, lease or order, or arranging for or recommending purchase, lease or order, of any good or service for which payment may be made under a federal healthcare program, such as Medicare and Medicaid; |
| • | the federal False Claims Act, or FCA, imposes criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
| • | the federal Health Insurance Portability and Accountability Act, or HIPAA, imposes criminal liability for knowingly and willfully executing a scheme to defraud any healthcare benefit program, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense or knowingly and willfully making false statements relating to healthcare matters; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 and its implementing regulations, also imposes obligations on certain covered entity healthcare providers, health plans and healthcare clearinghouses, and their business associates that perform certain services involving the use or disclosure of individually identifiable health information as well as their covered subcontractors, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| • | the federal Open Payments program, created under Section 6002 of the ACA and its implementing regulations, requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the United States Department of Health and Human Services, or HHS, information related to “payments or other transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals and applicable manufacturers and applicable group purchasing organizations to report annually to the HHS ownership and investment interests held by physicians (as defined above) and their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report such information regarding its relationships with physician assistants, nurse practitioners, anesthesiologist assistants, clinical nurse specialists, certified registered nurse anesthetists and certified nurse midwives during the previous year; and |
| • | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; state and foreign laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; state and local laws requiring the registration of pharmaceutical sales representatives; state and foreign laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, marketing expenditures or pricing; and state and foreign laws that govern the privacy and security of health information in certain circumstances, many of which differ from each |
S-37
Table of Contents
| other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, additional regulatory oversight, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other healthcare providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, that person or entity may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Our operations are vulnerable to interruption by fire, earthquake, power loss, telecommunications failure, terrorist activity and other events beyond our control, which could harm our business.
Our former facility was subject to electrical blackouts as a result of a shortage of available electrical power. Future blackouts, which may be implemented by the local electricity provider in the face of high winds and dry conditions, could disrupt the operations of our current facility. Our facility is located in a seismically active region. We have not undertaken a systematic analysis of the potential consequences to our business and financial results from a major earthquake, fire, power loss, terrorist activity or other disasters and do not have a complete recovery plan for such disasters. In addition, we do not carry sufficient insurance to compensate us for actual losses from interruption of our business that may occur, and any losses or damages incurred by us could harm our business. Our sole supplier of clinical drug substance for MK-3655 (NGM313), NGM120, NGM621, NGM707 and NGM438 is located in a region that has experienced recent political unrest.
Our business is currently adversely affected and could be materially and adversely affected in the future by the effects of disease outbreaks, epidemics and pandemics, including by the evolving effects of the COVID-19 pandemic. The COVID-19 pandemic continues to adversely impact our business and could materially and adversely affect our operations, as well as the businesses or operations of our manufacturers, CROs or other third parties with whom we conduct business.
Disease outbreaks and epidemics in regions where we have concentrations of clinical trial sites or other business operations or pandemics such as the COVID-19 pandemic could adversely affect our business, including by causing significant disruptions in our operations and/or in the operations of third-party manufacturers and CROs upon whom we rely. For example, the COVID-19 pandemic has presented a substantial public health and economic challenge around the world and is affecting employees, patients, communities and business operations, as well as the United States and international economy and financial markets. In this regard, the COVID-19 pandemic and government measures taken in response have had a significant impact, both direct and indirect, on businesses and commerce, as significant reductions in business-related activities have occurred, supply chains have been disrupted and manufacturing and clinical development activities have been curtailed or suspended.
Remote work policies, quarantines, shelter-in-place and similar government orders, shutdowns or other restrictions on the conduct of business operations related to the COVID-19 pandemic could
S-38
Table of Contents
materially and adversely affect our operations. Based on guidance issued by federal, state and local authorities, we transitioned to a remote work environment for a vast majority of our employees in March 2020, while maintaining essential in-person laboratory functions in order to advance key research and development initiatives, supported by the implementation of updated onsite safety procedures. In June 2020, following updated guidance issued by federal, state and local authorities, we re-opened our laboratory facilities for research activities that cannot be conducted remotely with heightened safety measures designed to minimize occupational exposure and reduce transmission of COVID-19 within our workplace. Although we have re-opened our laboratory facilities under these heightened safety measures, we may be forced to, or determine that we should, resume a more restrictive remote work model. In connection with these measures, we may be subject to claims based upon, arising out of, or related to COVID-19 and our actions and responses thereto, including any determinations that we have made and may make in the future with respect to our onsite operations. Further, the effects of current and future governmental shelter-in-place orders and our remote work policies may materially and adversely impact productivity, disrupt our business and delay our clinical programs and timelines, the magnitude of which will depend, in part, on the length and severity of the restrictions and other limitations on our ability to conduct our business in the ordinary course. These and similar, and perhaps more severe, disruptions in our operations could materially and adversely impact our business, financial condition, results of operations and growth prospects.
As the pandemic continues, there may be continuing negative impacts on our ability to initiate new clinical trial sites, maintain enrollment of existing patients and enroll new patients, which may impact timelines in the future. Our ability to attract additional clinical trial sites and principal investigators to conduct our clinical trials and to conduct the necessary clinical trial site initiation procedures has been and may continue to be impacted by quarantines, shelter-in-place and similar restrictions imposed by federal, state and local governments. These restrictions may also continue to prohibit or discourage patients from enrolling in, or continuing to participate in, our clinical trials. Principal investigators and clinical trial site staff, as healthcare providers, may have heightened exposure to COVID-19 and if their health is impacted by COVID-19 it could adversely impact the conduct of our clinical trials at their sites. Similarly, potential participants in our clinical trials, many of whom are particularly vulnerable, may be unwilling to enroll in, and enrolled patients may be unwilling to continue to participate in, our clinical trials due to concerns about traveling to sites for required screening and clinical trial visits and procedures. In this regard, we have experienced, from to time, a slower pace of clinical site initiation and enrollment than anticipated in certain of our clinical trials, including the ALPINE 4, CATALINA and NGM120 trials, and may experience higher dropout rates than normal, due to factors such as the vulnerability of our studied patient populations, clinical trial site suspensions, reallocation of medical resources and the challenges of working remotely due to shelter-in-place and similar government orders, among other factors. Enrolled patients may be unable to comply with clinical trial protocols if quarantines, shelter-in-place and similar restrictions continue to impede patient movement or interrupt healthcare services. Accordingly, we have developed and implemented additional clinical study policies and procedures designed to help protect patients from COVID-19 exposure as a result of their trial participation, which include the use of telemedicine visits, remote monitoring of patients and clinical trial sites, and other measures designed to ensure that data from clinical trials that may be disrupted as result of the pandemic are collected pursuant to the study protocol and consistent with current Good Clinical Practices, or cGCPs, with any material protocol deviation reviewed and approved by the clinical trial site IRB. If any of the foregoing efforts to mitigate the impact of the COVID-19 pandemic are not successful, or if the effects of the COVID-19 pandemic become more severe, it could materially and adversely affect our clinical development timelines and our ability to obtain regulatory approvals of our product candidates and could significantly increase our costs.
We could also see an adverse impact on our ability to report clinical trial results, or interact with regulators, IRBs and ethics committees or other important agencies due to limitations in regulatory
S-39
Table of Contents
authority employee resources or otherwise. Moreover, we rely on CROs and other third parties to assist us with clinical development activities, and we cannot guarantee that they will continue to perform their contractual duties in a timely and satisfactory manner as a result of the COVID-19 pandemic.
In addition, quarantines, shelter-in-place and similar government orders could impact personnel at third-party manufacturing facilities in the United States and other countries, or the availability or cost of materials, which would disrupt our supply chain. Likewise, the operations of our third-party manufacturers may be requisitioned, diverted or allocated by U.S. or foreign government orders such as under emergency, disaster and civil defense declarations in connection with the COVID-19 pandemic or otherwise. In particular, some of our suppliers of certain materials used in the production of our drug products are located in Europe. In this regard, any manufacturing supply interruption of aldafermin, which is currently manufactured by Lonza at facilities in Switzerland, or our other product candidates, which are currently manufactured at a facility in Lithuania, could adversely affect our ability to conduct ongoing and future clinical trials of aldafermin and our other product candidates. For example, although significant portions of our research and development resources are focused, and will continue to be focused, on activities required to prepare aldafermin for potential regulatory approval for the treatment of NASH, including manufacturing of clinical trial material and preparation for potential Phase 3 testing, if Lonza and/or our drug product manufacturer experience difficulties in scaling production or experience product loss due to contamination, equipment failure, improper installation or operation of equipment, vendor or operator error or improper storage conditions, the potential Phase 3 testing of aldafermin would be delayed, perhaps substantially, which could materially and adversely affect our business. Moreover, our aldafermin drug product manufacturer has advised us that it could be required under orders of the U.S. government to allocate manufacturing capacity to the manufacture or distribution of COVID-19 vaccines. If Lonza Sales AG and/or our aldafermin drug product manufacturer become subject to acts or orders of U.S. or foreign government entities to allocate manufacturing capacity to the manufacture or distribution of COVID-19 vaccines or medical supplies needed to treat COVID-19 patients, this could also delay, perhaps substantially, the potential Phase 3 testing of aldafermin which could materially and adversely affect our business. In any event, if the evolving effects of the COVID-19 pandemic become more severe or more acutely impact geographies with particular relevance to our business, we could experience significant disruptions to our current and potential future clinical development timelines, impacts on our ability to obtain regulatory approvals of our product candidates and increases in our costs, all or any of which would adversely affect our business, financial condition, results of operations and growth prospects.
In addition, while the potential economic impact brought by, and the duration of, the COVID-19 pandemic may be difficult to assess or predict, the pandemic could result in significant and prolonged disruption of global financial markets, reducing our ability to access capital, which could in the future negatively affect the financial resources available to us. In addition, the current recession or additional market corrections resulting from, among other things, the spread of COVID-19 could materially affect our business and the value of our common stock. We also cannot predict how the evolving effects of the COVID-19 pandemic may influence the future decisions of Merck to license any programs available to it under the Collaboration Agreement or to exercise its remaining option to extend the research phase of the collaboration beyond March 16, 2022.
While we expect the COVID-19 pandemic to continue to affect our business operations, the extent of the impact on our clinical development and regulatory efforts, our ability to raise sufficient additional capital on acceptable terms, if at all, the decisions of Merck and the value of and market for our common stock will depend on future developments that are highly uncertain and cannot be predicted with confidence at this time. Such developments include the ultimate duration and severity of the pandemic, travel restrictions, quarantines, social distancing and business closure requirements in the United States and in other countries, and the effectiveness of actions taken globally to contain and
S-40
Table of Contents
treat COVID-19. In addition, to the extent the evolving effects of the COVID-19 pandemic adversely affects our business and results of operations, it may also have the effect of heightening many of the other risks and uncertainties described elsewhere in this ‘‘Risk Factors’’ section.
Our internal computer systems, or those used by our CROs or other contractors or consultants, may fail or suffer security breaches.
Similar to other companies in our industry, we face substantial cybersecurity risk. Despite the implementation of security measures, our internal computer systems and those of our current and future CROs and other contractors, collaborators and consultants may fail and are vulnerable to damage from computer viruses and unauthorized access. While we have not, to our knowledge, experienced any material system failure or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Likewise, we rely on third parties for the manufacture of our product candidates, to analyze clinical trial samples and to conduct clinical trials, and similar events relating to their computer systems could also have a material adverse effect on our business.
As a result of the ongoing COVID-19 pandemic, certain functional areas of our workforce remain in a remote work environment, which imposes additional risks to our business, including increased risk associated with working outside our corporate network security protection boundaries, increased risk of industrial espionage, phishing and other cybersecurity attacks and the increased risk of unauthorized dissemination of sensitive personal information or proprietary confidential information, any of which could have a material adverse effect on our business. Despite our efforts to increase security and authentication measures, we have experienced an overall increase in cybersecurity incidents, none of which have caused disruption to our business or resulted in a material security breach. However, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations.
In 2017, a security breach of the internal computer systems of our collaborator, Merck, caused material damage to its operations, but did not affect our internal operations. In June 2019, a vendor that conducted bioanalytical services for some of our aldafermin clinical trials was affected by a ransomware attack that resulted in a significant disruption to its IT systems. This cybersecurity incident at our vendor did not result in an integrity loss of certain clinical sample data for aldafermin, as verified by independent vendors. More recently, an attacker gained access to a single system on our network and attempted to use that access to stage a broader attack against us. We detected the suspicious activity on the night of December 15, 2020 and believe we fully contained the incident the next day. The event had minimal impact on our operations and we have no evidence suggesting any ongoing threat or data exfiltration. However, to the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur material costs, be exposed to liability, our competitive position could be harmed and the further development and commercialization of our product candidates could be hindered or delayed.
European data collection is governed by restrictive regulations governing the collection, use, processing and cross-border transfer of personal information.
We may collect, process, use or transfer personal information from individuals located in the EU in connection with our business, including in connection with conducting clinical trials in the EU. Additionally, if any of our product candidates are approved, we may seek to commercialize those
S-41
Table of Contents
products in the EU. The collection and use of personal health data in the EU are governed by the provisions of the General Data Protection Regulation ((EU) 2016/679), or GDPR. This legislation imposes requirements relating to having legal bases for processing personal information relating to identifiable individuals and transferring such information outside of the European Economic Area, including to the United States, providing details to those individuals regarding the processing of their personal information, keeping personal information secure, having data processing agreements with third parties who process personal information, responding to individuals’ requests to exercise their rights in respect of their personal information, reporting security breaches involving personal data to the competent national data protection authority and affected individuals, appointing data protection officers, conducting data protection impact assessments and record-keeping. The GDPR imposes additional responsibilities and liabilities in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with the new data protection rules. Failure to comply with the requirements of the GDPR and related national data protection laws of the member states of the EU may result in substantial fines, other administrative penalties and civil claims being brought against us, which could have a material adverse effect on our business, financial condition and results of operations.
European data protection laws, including the GDPR, generally restrict the transfer of personal information from Europe, including the European Economic Area, United Kingdom and Switzerland, to the United States and most other countries unless the parties to the transfer have implemented specific safeguards to protect the transferred personal information. One of the primary safeguards allowing United States companies to import personal information from Europe has been certification to the EU-U.S. Privacy Shield and Swiss-U.S. Privacy Shield frameworks administered by the United States Department of Commerce. However, the Court of Justice of the EU recently invalidated the EU-U.S. Privacy Shield. The same decision also raised questions about whether one of the primary alternatives to the EU-U.S. Privacy Shield, namely, the European Commission’s Standard Contractual Clauses, can lawfully be used for personal information transfers from Europe to the United States or most other countries. At present, there are few, if any, viable alternatives to the EU-U.S. Privacy Shield and the Standard Contractual Clauses. Although we rely primarily on individuals’ explicit consent to transfer their personal information from Europe to the United States and other countries, in certain cases we have relied or may rely on the Standard Contractual Clauses. Authorities in the United Kingdom and Switzerland, whose data protection laws are similar to those of the EU, may similarly invalidate use of the EU-U.S. Privacy Shield and Swiss-U.S. Privacy Shield, respectively, as mechanisms for lawful personal information transfers from those countries to the United States. As such, if we are unable to rely on explicit consent to transfer individuals’ personal information from Europe, which can be revoked, or implement another valid compliance solution, we will face increased exposure to substantial fines under European data protection laws as well as injunctions against processing personal information from Europe. Inability to import personal information from the European Economic Area, United Kingdom or Switzerland may also restrict our clinical trial activities in Europe; limit our ability to collaborate with CROs, service providers, contractors and other companies subject to European data protection laws; and require us to increase our data processing capabilities in Europe at significant expense. Additionally, other countries outside of Europe have enacted or are considering enacting similar cross-border data transfer restrictions and laws requiring local data residency, which could increase the cost and complexity of delivering our services and operating our business.
We use and generate materials that may expose us to material liability.
Our research programs involve the use of hazardous materials, chemicals and radioactive and biological materials. We are subject to foreign, federal, state and local environmental and health and safety laws and regulations governing, among other matters, the use, manufacture, handling, storage and disposal of hazardous materials and waste products. We may incur significant costs to comply with these current or future environmental and health and safety laws and regulations. In addition, we
S-42
Table of Contents
cannot completely eliminate the risk of contamination or injury from hazardous materials and may incur material liability as a result of such contamination or injury. In the event of an accident, an injured party may seek to hold us liable for any damages that result. Any liability could exceed the limits or fall outside the coverage of our workers’ compensation, property and business interruption insurance and we may not be able to maintain insurance on acceptable terms, if at all. We currently carry no insurance specifically covering environmental claims.
Risks Related to Our Dependence on Merck and Other Third Parties
We depend on our collaboration with Merck and may depend in the future on collaborations with additional third parties for the development and commercialization of our product candidates. If those collaborations are not successful, we may not be able to capitalize on the market potential of our product candidates.
In February 2015, we entered into a collaboration with Merck focused on the discovery, development and commercialization of biologics, excluding aldafermin, and including a license to our GDF15 receptor agonist program product candidates, NGM386 and NGM395. In November 2018, Merck exercised its option to license MK-3655 (NGM313). In 2019, Merck exercised its option to extend the research phase of the collaboration for an additional two years, from March 17, 2020 through March 16, 2022, and terminated its license to the GDF15 receptor agonist program in May 2019.
The Merck collaboration involves a complex allocation of rights, provides for substantial research and development support, provides for additional payments upon Merck’s election, if exercised in its unilateral discretion, to further extend the term of the research program for an additional two years and provides us with either milestone payments based on the achievement of specified clinical development, regulatory and commercial milestones and royalty-based revenue if certain product candidates are successfully commercialized or a cost and profit sharing arrangement with the possibility that we would provide sales representatives to co-detail the product candidates that Merck elects to advance in the United States. We cannot predict the success of the collaboration, whether Merck will exercise its remaining option to extend the research phase of the collaboration or whether Merck will exercise its option to license additional product candidates or whether Merck will terminate its license to a licensed program.
We may seek other third-party collaborators for the development and commercialization of any product candidates that are not subject to the Merck collaboration, including aldafermin, NGM395 and NGM386. Our likely collaborators for any collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies and biotechnology companies. If we enter into any such arrangements with any third parties, we will likely have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidates. Our ability to generate revenue from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
Collaborations involving our product candidates, including our collaboration with Merck, pose risks to us, including the following:
| • | Collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations. For example, under our collaboration with Merck, once proof-of-concept data in humans has been generated and Merck has exercised its option to acquire an exclusive license for a product candidate, our ability to influence the resources Merck devotes to such product candidate are substantially reduced until such time, if any, that |
S-43
Table of Contents
| we exercise our right to participate in a cost and profit-sharing arrangement. Even after we exercise that right to participate in a cost and profit-sharing arrangement, our ability to influence Merck will be limited. |
| • | Collaborators might opt not to pursue development and commercialization of our product candidates or not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborator’s strategic focus or available funding or external factors, such as an acquisition that diverts resources or creates competing priorities. For example, under our agreement with Merck, it is possible for Merck to unilaterally terminate the MK-3655 (NGM313) program and any other program (whether or not we have exercised our cost and profit-sharing option) upon prior written notice, such as it did for NGM386 and NGM395, without triggering a termination of the remainder of the collaboration arrangement. In addition, Merck might opt not to exercise its option to acquire a license to a product candidate that has generated proof-of-concept data. |
| • | Collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing. |
| • | Collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our medicines or product candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours. |
| • | A collaborator with marketing and distribution rights to one or more medicines might not commit sufficient resources to the marketing and distribution of our product candidates. |
| • | Collaborators might not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our proprietary information or expose us to potential litigation. For example, Merck has the first right to maintain or defend our intellectual property rights under the Collaboration Agreement with respect to certain licensed programs and, although we may have the right to assume the maintenance and defense of our intellectual property rights if Merck does not, our ability to do so may be compromised by Merck’s actions. |
| • | Disputes may arise between the collaborators and us that result in the delay or termination of the research, development or commercialization of our medicines or product candidates or that result in costly litigation or arbitration that diverts management attention and resources. |
| • | We may lose certain valuable rights under circumstances identified in our collaborations, including, in the case of our agreement with Merck, if we undergo a change in control. |
| • | Collaborations might be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates. |
| • | Collaboration agreements might not lead to development or commercialization of product candidates in the most efficient manner, or at all. If a present or future collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program under such collaboration could be delayed, diminished or terminated. |
Under certain circumstances, Merck may unilaterally terminate its annual funding of our research and development program, or terminate or shift the focus of its research and development funding, any of which would materially and adversely affect our business.
Under the Collaboration Agreement, Merck has the unilateral right to terminate all or part of the agreement at certain times and under certain circumstances. Merck may unilaterally terminate the
S-44
Table of Contents
research phase of the collaboration program effective March 17, 2022 by providing notice to us prior to March 17, 2021. Merck may also unilaterally terminate its annual funding of the research program prior to March 17, 2022 if we are acquired by a third party or if we are in material uncured breach of our obligations under the research and early development program. After the current research phase of the collaboration or, if Merck again exercises its option to extend the research phase of the collaboration, after such extension period, Merck may unilaterally terminate the overall agreement for convenience upon written notice and subject to certain limitations.
Subject to certain limitations, Merck may partially terminate the Collaboration Agreement for convenience as it relates to MK-3655 (NGM313) or any future licensed program. For example, Merck terminated its license to our GDF15 receptor agonist program, including NGM395 and NGM386, in May 2019. Merck may also unilaterally terminate the agreement as it relates to its rights to research and develop small molecule compounds. It may also unilaterally terminate the agreement with respect to a specific licensed program in the event of an uncured material breach by us. If Merck terminates a program as a result of our uncured material breach, then we would lose our option to participate in global cost and profit sharing if not yet exercised as of the time of termination, and lose our co-detailing option (whether or not exercised as of that time) for the relevant licensed program.
If Merck terminates funding, terminates the Collaboration Agreement, decides not to further extend the research phase of the collaboration or shifts the focus of its research and development funding, it would delay or preclude our ability complete our research and development programs, which would materially and adversely affect our business. For example, if Merck elects not to exercise its remaining option to extend the research phase of the collaboration beyond March 16, 2022, which Merck may unilaterally elect to do at its sole discretion, we would require significant additional capital in order to proceed with development and commercialization of any product candidates that had been subject to the Merck collaboration but Merck decides not to proceed with after termination of the research phase, or we will need to enter into additional collaboration or license agreements in order to fund such development and commercialization, which may not be possible, or we may be required to delay, scale back or discontinue development of such product candidates.
We may not be able to obtain and maintain the relationships with third parties that are necessary to develop, commercialize and manufacture some or all of our product candidates.
In addition to our dependence on our collaboration with Merck, we expect to depend on other collaborators, partners, licensees, CROs, clinical investigators, manufacturers and other third parties to support our discovery efforts, to formulate product candidates, to conduct clinical trials for some or all of our product candidates, to manufacture clinical and commercial scale quantities of our drug substances and drug products and to market, sell and distribute any products we successfully develop and for which we obtain regulatory approval. Any problems we experience with any of these third parties could delay our research efforts or the development, commercialization and manufacturing of our products or product candidates, which could harm our results of operations.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured product candidates or products ourselves, including reliance on third parties for regulatory compliance and quality assurance, the possibility of breach of the manufacturing agreement by third parties because of factors beyond our control (including a failure to manufacture our product candidates or any products we may eventually commercialize in accordance with our specifications) and the possibility of termination or nonrenewal of agreements by third parties, based on its own business priorities, at a time that is costly or damaging to us.
If any of our product candidates are approved by the FDA or other regulatory agencies for commercial sale, we or our collaborator may need to manufacture it in larger quantities. We intend to use third-party manufacturers for commercial quantities of aldafermin, NGM120, NGM621, NGM707
S-45
Table of Contents
and NGM438, to the extent we advance these product candidates, and will rely on Merck to determine whether to utilize a third-party manufacturer or internal manufacturing capacity for MK-3655 (NGM313) and other licensed product candidates. Our or our collaborator’s manufacturers may not be able to successfully increase the manufacturing capacity for any of our product candidates in a timely or efficient manner, or at all. If we or our collaborator are unable to successfully increase the manufacturing capacity for a product candidate, the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage in the supply of the product candidate. Our or our collaborator’s failure or the failure of third-party manufacturers to comply with the FDA’s current Good Manufacturing Practices, or cGMP, and to pass inspections of the manufacturing facilities by the FDA or other regulatory agencies could seriously harm our business.
We cannot guarantee that we or, as applicable, our collaborator will be able to successfully negotiate agreements for, or maintain relationships with, collaborators, partners, licensees, CROs, clinical investigators, manufacturers and other third parties on favorable terms, if at all. If we or our collaborator are unable to obtain or maintain these agreements, we may not be able to clinically develop, formulate, manufacture, obtain regulatory approvals for or commercialize our product candidates, which will, in turn, adversely affect our business.
We expect to expend substantial management time and effort to enter into relationships with third parties and, if we successfully enter into such relationships, to manage these relationships. In addition, substantial capital will be paid to third parties in these relationships. However, we cannot control the amount or timing of resources our contract partners will devote to our research and development programs, product candidates or potential product candidates, and we cannot guarantee that these parties will fulfill their obligations to us under these arrangements in a timely fashion, if at all. In addition, we are less knowledgeable about the reputation and quality of third-party contractors in countries outside of the United States where we conduct discovery research or preclinical and clinical development and manufacturing of our product candidates and, therefore, we may not choose the best parties for these relationships.
We may seek to establish additional collaborations, and, if we are not able to establish them on commercially reasonable terms, we may have to alter our development and commercialization plans.
Our drug development programs and the potential commercialization of our product candidates will require substantial additional cash to fund expenses. For product candidates not partnered with Merck, such as aldafermin, NGM395 and NGM386, we may decide to collaborate with other pharmaceutical and biotechnology companies for their development and potential commercialization.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA or comparable foreign regulatory authorities, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products and the existence of uncertainty with respect to our ownership of intellectual property, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. Potential collaborators may also consider alternative product candidates or intellectual property for similar indications that may be available for collaboration and whether such an alternative collaboration could be more attractive than the one with us for our product candidate.
S-46
Table of Contents
We may also be restricted under existing collaboration agreements from entering into future agreements on certain terms with potential collaborators. For example, under our Collaboration Agreement with Merck, we may not directly or indirectly research, develop, manufacture or commercialize, except pursuant to the Collaboration Agreement, any medicine or product candidate that modulates a target then subject to the collaboration with specified activity. The FGF19 program, including aldafermin, is excluded from this provision, notwithstanding that both aldafermin and MK-3655 (NGM313) signal, in part, through the FGFR1c pathway. During the tail period following the research term of the collaboration, we may not directly or indirectly research, develop or commercialize, outside of the collaboration, any medicine or product candidate with specified activity against any collaboration target that has been designated a tail target.
Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
We may not be able to negotiate collaborations on a timely basis, on acceptable terms or at all. If we are unable to do so, we may have to delay, scale back or discontinue the development of any product candidate for which we are seeking a collaboration, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms, or at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate product revenue.
We rely on third parties to conduct our clinical trials and some aspects of our research and preclinical studies, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials, research or testing.
We currently rely on, and expect to continue to rely on, third parties, such as CROs, clinical data management organizations, medical institutions, consultants and clinical investigators, to conduct our clinical trials and certain aspects of our research and preclinical studies. Any of these third parties may terminate their engagements with us at any time. If we need to enter into alternative arrangements, it would delay our product development activities and such alternative arrangements may not be available on terms acceptable to us.
Our reliance on these third parties for research and development activities will reduce our control over these activities, but will not relieve us of our responsibilities. For example, we will remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with standards, commonly referred to as cGCPs, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, ClinicalTrials.gov, within certain timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully
S-47
Table of Contents
commercialize our medicines. In this regard, we cannot guarantee that these third parties will continue to perform their contractual duties in a timely and satisfactory manner as a result of the COVID-19 pandemic or otherwise.
We also expect to rely on third parties to store and distribute drug supplies for our clinical trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our product candidates or commercialization of our medicines, producing additional losses and depriving us of potential product revenue.
In addition, we rely on these third parties to provide accurate financial information related to our research and development activities and if any inaccurate financial information were provided by these third parties, our results of operations could be adversely impacted.
We contract with third parties for the manufacture of our product candidates for preclinical and clinical studies and expect to continue to do so for commercialization. This reliance on third parties increases the risk that we will not have sufficient quantities of our product candidates or that such supply will not be available to us on our requested timeline or at an acceptable cost, which could delay, prevent or impair our development or commercialization efforts.
We do not have any manufacturing facilities. We currently rely, and expect to continue to rely, on third-party manufacturers for the manufacture of our product candidates for preclinical and clinical studies and for commercial supply of any of these product candidates for which we or our collaborator obtains marketing approval. To date, we have obtained aldafermin, MK-3655 (NGM313), NGM120, NGM621, NGM707 and NGM438 for preclinical and clinical studies from third-party manufacturers. Other than our supply agreement with Lonza for aldafermin drug substance, we do not have a long-term supply agreement with any third-party manufacturer.
We may be unable to establish any agreements with third-party manufacturers on acceptable terms or at all. Even if we are able to establish agreements with third-party manufacturers, reliance on third-party manufacturers entails additional risks, including:
| • | the possible breach of the manufacturing agreement by the third party or the failure to supply product on the timelines and at the cost agreed to; |
| • | the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us; and |
| • | reliance on the third party for regulatory compliance, quality assurance and safety and pharmacovigilance reporting. |
Third-party manufacturers may not be able to comply with cGMP or similar regulatory requirements outside the United States. Our failure, or the failure of third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or medicines, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our medicines and harm our business and results of operations.
Any medicines that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing our product candidates.
S-48
Table of Contents
Any performance failure on the part of our existing or future manufacturers could delay clinical development or marketing approval. In this regard, remote work policies and quarantines, shelter-in-place and similar government orders related to the COVID-19 pandemic could impact personnel at third-party manufacturing facilities in the United States and other countries, or the availability or cost of materials, which would disrupt our supply chain and delay our clinical development efforts. In particular, some of our suppliers of certain materials used in the production of our drug products are located in Europe. For example, any manufacturing supply interruption of aldafermin, which is currently manufactured by Lonza at facilities in Switzerland, or our other product candidates, which are currently manufactured at a facility in Lithuania, could adversely affect our ability to conduct ongoing and future clinical trials of aldafermin and our other product candidates. We do not currently have arrangements in place for redundant supply for bulk drug substances or drug product. If any one of our current contract manufacturers cannot perform as agreed, we may be required to replace that manufacturer, if possible. Although there may be potential alternative manufacturers who could manufacture our product candidates, we would incur additional costs and experience delays in identifying and qualifying any such replacement.
Our current and anticipated future dependence upon others for the manufacture of our product candidates or any future products may adversely affect our future profit margins and our ability to commercialize any products that receive marketing approval on a timely and competitive basis. Refer also to the risk factor entitled “We are subject to many manufacturing risks, any of which could substantially increase our costs and limit supply of our product candidates and any future products.”
Risks Related to Regulatory Approvals
None of our product candidates has received regulatory approval. If we are unable to obtain regulatory approvals to market one or more of our product candidates, our business will be adversely affected.
We do not expect our product candidates to be commercially available for several years, if at all. Our product candidates are subject to strict regulation by regulatory authorities in the United States and in other countries. We cannot market any product candidate until we have completed all necessary preclinical studies and clinical trials and have obtained the necessary regulatory approvals. The approval process is typically lengthy and expensive, and approval is never certain. We do not know whether regulatory agencies will grant approval for any of our product candidates. Even if we complete preclinical studies and clinical trials successfully, we may not be able to obtain regulatory approvals or we may not receive approvals to make claims about our products that we believe to be necessary to effectively market our products. Data obtained from preclinical studies and clinical trials is subject to varying interpretations that could delay, limit or prevent regulatory approval, and failure to comply with regulatory requirements or inadequate manufacturing processes are examples of other problems that could prevent approval. In addition, we may encounter delays or rejections due to additional government regulation from future legislation, administrative action or changes in the policies of the FDA or comparable foreign regulatory authorities. Even if the FDA or a comparable foreign regulatory authority approves a product, the approval will be limited to those indications covered in the approval.
The regulatory approval processes of the FDA and comparable foreign regulatory authorities are lengthy, time-consuming and inherently unpredictable. Our inability to obtain regulatory approval for our product candidates would substantially harm our business.
Currently, none of our product candidates has received regulatory approval. The time required to obtain approval from the FDA and comparable foreign regulatory authorities is unpredictable but typically takes many years following the commencement of preclinical studies and clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations or the type and amount of preclinical and clinical data
S-49
Table of Contents
necessary to gain approval may change during the course of a product candidate’s development and may vary among jurisdictions. It is possible that none of our existing or future product candidates will ever obtain regulatory approval.
Our product candidates could fail to receive regulatory approval from the FDA or a comparable foreign regulatory authority for many reasons, including:
| • | disagreement with the design or implementation of our clinical trials; |
| • | failure to demonstrate that a product candidate is safe and effective for its proposed indication; |
| • | failure of results of clinical trials to meet the level of statistical significance required for approval; |
| • | failure to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
| • | disagreement with our interpretation of data from preclinical studies or clinical trials; |
| • | the insufficiency of data collected from clinical trials to support the submission and filing of a BLA or other submission or to obtain regulatory approval; |
| • | failure to obtain approval of the manufacturing processes or facilities of third-party manufacturers with whom we contract for clinical and commercial supplies; or |
| • | changes in the approval policies or regulations that render our preclinical and clinical data insufficient for approval. |
The FDA or a comparable foreign regulatory authority may require more information, including additional preclinical or clinical data, to support approval, which may delay or prevent approval and commercialization, or we may decide to abandon the development program for other reasons. If we obtain approval, regulatory authorities may approve any of our product candidates for fewer or more limited indications than we request, may grant approval contingent on the performance of costly post-marketing clinical trials or may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate.
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drug products that meet certain criteria. Specifically, new drugs are eligible for Fast Track designation if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition, and the FDA may grant accelerated approval based on a surrogate endpoint reasonably likely to predict clinical benefit. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. Unique to a Fast Track product, the FDA may consider sections of the BLA for review on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the BLA, the FDA agrees to accept sections of the BLA and determines that the schedule is acceptable and the sponsor pays any required user fees upon submission of the first section of the BLA. Fast Track designation does not change the standards for product approval.
Although aldafermin has received Fast Track designation from the FDA for NASH and PBC, we may not necessarily experience faster development timelines or achieve faster review or approval compared to conventional FDA procedures. Many agents in development for NASH have, or are expected to, opt for an accelerated approval pathway and rely on surrogate endpoints for initial approval. If we seek accelerated approval for one of our product candidates, including aldafermin for NASH, based on a surrogate endpoint, the FDA may not accept such endpoint, may require additional studies or analysis or may not approve our product candidate on an accelerated basis, or at all. For example, in June 2020, Intercept announced that it had received a complete response letter regarding
S-50
Table of Contents
its New Drug Application for OCA for the treatment of NASH, in which the FDA indicated that it had determined that the predicted benefit of OCA based on a surrogate histopathologic endpoint was uncertain and did not sufficiently outweigh the potential risks to support accelerated approval for the treatment of patients with liver fibrosis due to NASH. The FDA recommended that Intercept submit additional post-interim analysis efficacy and safety data from its ongoing Phase 3 study in support of potential accelerated approval and that the long-term outcomes phase of the study should continue.
Further, access to an expedited program may also be withdrawn by the FDA if it believes that the designation is no longer supported by data from our clinical development program. Additionally, qualification for any expedited review procedure does not ensure that we will ultimately obtain regulatory approval for aldafermin or any other product candidate that we are developing or may develop.
Our failure to obtain regulatory approval in international jurisdictions would prevent us from marketing our product candidates outside the United States.
If we or our collaborators succeed in developing any products, we intend to market them in the EU and other foreign jurisdictions in addition to the United States. In order to market and sell our products in other jurisdictions, we must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA approval. The regulatory approval process outside the United States generally includes all of the risks associated with obtaining FDA approval. In addition, in many countries outside the United States, we must secure product reimbursement approvals before regulatory authorities will approve the product for sale in that country. Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our products in certain countries. Further, clinical trials conducted in one country may not be accepted by regulatory authorities in other countries and regulatory approval in one country does not ensure approval in any other country, while a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory approval process in others. Also, regulatory approval for any of our product candidates may be withdrawn if we fail to comply with regulatory requirements, if problems occur after the product candidate reaches the market or for other reasons. If we fail to comply with the regulatory requirements in international markets and fail to receive applicable marketing approvals, our target market will be reduced and our ability to realize the full market potential of our product candidates will be harmed and our business will be adversely affected. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions. Approval by one regulatory authority outside the United States does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. If we fail to obtain approval of any of our product candidates by regulatory authorities in another country, we will be unable to commercialize our product in that country, and the commercial prospects of that product candidate and our business prospects could decline.
Even if our product candidates receive regulatory approval, they may still face future development and regulatory difficulties.
Even if we obtained regulatory approval for a product candidate, it would be subject to ongoing requirements by the FDA and comparable foreign regulatory authorities governing the manufacture, quality control, further development, labeling, packaging, storage, distribution, safety surveillance, import, export, advertising, promotion, recordkeeping and reporting of safety and other post-market information. The FDA and comparable foreign regulatory authorities will continue to closely monitor the safety profile of any product even after approval. If the FDA or comparable foreign regulatory authorities become aware of new safety information after approval of any of our product candidates,
S-51
Table of Contents
they may require labeling changes or establishment of a Risk Evaluation and Mitigation Strategy or similar strategy, impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance.
In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP regulations. If we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. If we, our product candidates or the manufacturing facilities for our product candidates fail to comply with applicable regulatory requirements, or undesirable side effects caused by such products are identified, a regulatory agency may:
| • | issue safety alerts, Dear Healthcare Provider letters, press releases or other communications containing warnings about such product; |
| • | mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners; |
| • | require that we conduct post-marketing studies; |
| • | require us to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
| • | seek an injunction or impose civil or criminal penalties or monetary fines; |
| • | suspend marketing of, withdraw regulatory approval of or initiate a recall of such product; |
| • | suspend any ongoing clinical trials; |
| • | refuse to approve pending applications or supplements to applications filed by us; |
| • | suspend or impose restrictions on operations, including costly new manufacturing requirements; or |
| • | seize or detain products or refuse to permit the import or export of products. |
The occurrence of any event or penalty described above may inhibit our ability to commercialize our products and generate revenue.
Advertising and promotion of any product candidate that obtains approval in the United States will be heavily scrutinized by the FDA, Department of Justice, HHS, Office of Inspector General, state attorneys general, members of Congress and the public. Violations, including promotion of our products for unapproved (or off-label) uses, are subject to enforcement letters, inquiries and investigations and civil and criminal sanctions by the government. Additionally, comparable foreign regulatory authorities will heavily scrutinize advertising and promotion of any product candidate that obtains approval outside of the United States.
In the United States, engaging in the impermissible promotion of our products for off-label uses can also subject us to false claims litigation under federal and state statutes, which can lead to civil and criminal penalties and fines and agreements that materially restrict the manner in which a company promotes or distributes drug products. These false claims statutes include the federal FCA, which allows any individual to bring a lawsuit against a pharmaceutical company on behalf of the federal government alleging submission of false or fraudulent claims, or causing to present such false or fraudulent claims, for payment by a federal program such as Medicare or Medicaid. If the government prevails in the lawsuit, the individual will share in any fines or settlement funds. Since 2004, these FCA lawsuits against pharmaceutical companies have increased significantly in volume and breadth,
S-52
Table of Contents
leading to several substantial civil and criminal settlements regarding certain sales practices promoting off-label drug uses involving fines in excess of $1 billion. This growth in litigation has increased the risk that a pharmaceutical company will have to defend a false claim action, pay settlement fines or restitution, agree to comply with burdensome reporting and compliance obligations and be excluded from Medicare, Medicaid and other federal and state healthcare programs. If we do not lawfully promote our approved products, we may become subject to such litigation and, if we do not successfully defend against such actions, those actions may have a material adverse effect on our business, financial condition and results of operations.
The FDA’s policies may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
Even if we are able to obtain regulatory approvals for any of our product candidates, if they exhibit harmful side effects after approval, our regulatory approvals could be revoked or otherwise negatively impacted, and we could be subject to costly and damaging product liability claims.
Even if we receive regulatory approval for aldafermin or any of our other product candidates, we will have tested them in only a small number of patients during our clinical trials. If our applications for marketing are approved and more patients begin to use our product, new risks and side effects associated with our products may be discovered. As a result, regulatory authorities may revoke their approvals. If aldafermin is approved by the FDA based on a surrogate endpoint pursuant to accelerated approval regulations (Subpart E regulations), we will be required to conduct additional clinical trials establishing clinical benefit on the ultimate outcome of NASH. Additionally, we may be required to conduct additional clinical trials, make changes in labeling of our product, reformulate our product or make changes and obtain new approvals for our and our suppliers’ manufacturing facilities for aldafermin and our other product candidates. We might have to withdraw or recall our products from the marketplace. We may also experience a significant drop in the potential sales of our product if and when regulatory approvals for such product are obtained, experience harm to our reputation in the marketplace or become subject to lawsuits, including class actions. Any of these results could decrease or prevent any sales of our approved product or substantially increase the costs and expenses of commercializing and marketing our product.
Risks Related to Our Intellectual Property
Our success depends upon our ability to obtain and maintain intellectual property protection for our products and technologies.
Our success depends in significant part on our ability and the ability of our current or future licensors, licensees or collaborators to establish and maintain adequate intellectual property covering the product candidates that we plan to develop. In addition to taking other steps to protect our intellectual property, we have applied for, and intend to continue applying for, patents with claims covering our technologies, processes and product candidates when and where we deem it appropriate to do so. However, the patent prosecution process is expensive and time-consuming, and we and our current or future licensors, licensees or collaborators may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we or our current or future licensors, licensees or collaborators will fail to identify patentable aspects of inventions made in the course of development and commercialization activities
S-53
Table of Contents
before it is too late to obtain patent protection for them. Pending and future patent applications filed by us or our current or future licensors’, licensees’ or collaborators’ may not result in patents being issued that protect our technology or product candidates, or products resulting therefrom, in whole or in part, or that effectively prevent others from commercializing competitive technologies and products.
We have filed numerous patent applications both in the United States and in certain foreign jurisdictions to obtain patent rights to our inventions, with claims directed to compositions of matter, methods of use, formulations, combination therapy and other technologies relating to our product candidates. However, patent applications cannot be enforced against third parties practicing the technology that is currently claimed in such applications unless and until a patent issues from such applications, and then only to the extent the claims that issue are broad enough to cover the technology being practiced by third parties. There can be no assurance that any of these patent applications will issue as patents or, for those applications that do mature into patents, whether the claims of the patents will exclude others from making, using or selling our product or product candidates, or products or product candidates that are substantially similar to ours. In countries where we have not and do not seek patent protection, third parties may be able to manufacture and sell products that are substantially similar or identical to our products or product candidates without our permission, and we may not be able to stop them from doing so.
Similar to other biotechnology companies, our patent position is generally highly uncertain and involves complex legal and factual questions. The patent examination process may require us or our current or future licensors, licensees or collaborators to narrow the scope of the claims of pending and future patent applications, which would limit the scope of patent protection that is obtained, if any. The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our patents may be challenged in the courts or patent offices in the United States and abroad. In recent years, these areas have been the subject of much litigation in the industry. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights and those of our current or future licensors, licensees or collaborators are highly uncertain and may not effectively prevent others from commercializing competitive technologies and products.
Success in obtaining and maintaining patents and other intellectual property rights may depend upon being the first to make a particular invention or being the first inventor to file a patent application on that invention. However, the publication of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing or, in some cases, not at all until they are issued as a patent. Therefore, we cannot be certain that we or our current or future licensors, licensees or collaborators were the first to make an invention, or the first inventors to file a patent application claiming an invention in our owned or licensed patents or pending patent applications.
In some circumstances, we may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain or enforce the patents, covering technology that we license from or license to third parties and may be reliant on our current or future licensors, licensees or collaborators to perform these activities, which means that these patent applications may not be prosecuted, and these patents may not be enforced, in a manner consistent with the best interests of our business. If our current or future licensors, licensees or collaborators fail to establish, maintain, protect or enforce such patents and other intellectual property rights, such rights may be reduced or eliminated. If our current or future licensors, licensees or collaborators are not fully cooperative or disagree with us as to the prosecution, maintenance or enforcement of any patent rights, such patent rights could be compromised.
We do not currently own or have a license to any issued patents that cover our NGM621 product candidate, although it is disclosed and claimed in our pending U.S. non-provisional and/or national
S-54
Table of Contents
stage applications in particular foreign countries. Likewise, we do not currently own or have a license to any issued patents that cover our NGM707 and NGM438 product candidates, although these product candidates are disclosed and claimed in our pending U.S. provisional applications. The patent landscape surrounding NGM621, NGM707 and NGM438 are crowded, and there can be no assurance that we will be able to secure patent protection that would adequately cover such product candidates, nor can there be any assurance that we would obtain sufficiently broad claims to be able to prevent others from selling competing products.
Any changes we make to our product candidates, including aldafermin, MK-3655 (NGM313), NGM120, NGM621, NGM707 and NGM 438, to cause them to have what we view as more advantageous properties may not be covered by our existing patents and patent applications, and we may be required to file new patent applications and/or seek other forms of protection for any such altered product candidates. The patent landscape surrounding the technology underlying our product candidates is crowded, and there can be no assurance that we would be able to secure patent protection that would adequately cover an alternative to our aldafermin molecule, including half-life extending formulation enhancements or the half-life extended variants of FGF19 that we are developing, MK-3655 (NGM313), NGM120, NGM621, NGM707 and NGM438 or any of our other product candidates.
We may not be able to protect our intellectual property rights throughout the world.
The legal protection afforded to inventors and owners of intellectual property in countries outside of the United States may not be as broad or effective as that in the United States and we may be unable to acquire and enforce intellectual property rights outside the United States to the same extent as in the United States, if at all. Thus, our existing patents and any future patents we obtain may not be sufficiently broad to prevent others from practicing our inventions or technologies, or from developing or commercializing competing products.
Filing, prosecuting, enforcing and defending patents on product candidates in all countries throughout the world is prohibitively expensive, and our intellectual property rights in some countries outside the United States are less extensive than those in the United States. Competitors may use our technology and that of our current or future licensors, licensees or collaborators in jurisdictions where we have not obtained patent protection to develop their own products and, in some cases, may export otherwise infringing products to territories where we and our collaborator have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and product candidates, and our patents or other intellectual property rights may not be effective or sufficient to prevent that competition.
The requirements for patentability differ in certain countries, particularly developing countries. For example, China has a heightened requirement for patentability and, specifically, requires a detailed description of medical uses of a claimed drug. Europe and other countries impose limitations on inventions related to methods for treating the human body. Differences in national law and practice may limit the breadth and strength of any patents obtained by us or our current or future licensors, licensees or collaborators.
India, certain countries in Europe, and certain developing countries, including Thailand, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. Additionally, many countries limit the enforceability of patents against government agencies or government contractors. We or our licensors, licensees or collaborators may have limited remedies if patents are infringed or if compelled to grant a license to a third party, which could materially diminish the value of those patents and could limit our potential revenue opportunities. Accordingly, our efforts, and those of our licensors, licensees or collaborators, to enforce intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we own or license.
S-55
Table of Contents
Patent applications, issued patents and/or parts thereof must be translated into the native language in many foreign countries. If our patent applications or issued patents are translated incorrectly, they may not adequately cover our products, product candidates, or other technologies and it may not be possible to rectify an incorrect translation.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals. This could make it difficult for us or our current or future licensors, licensees or collaborators to stop the infringement of the patents or the marketing of competing products in violation of the patent or other proprietary rights. Proceedings to enforce patent rights in foreign jurisdictions could result in substantial costs and divert the attention of us or our current or future licensors, licensees or collaborators from other aspects of our business, could put the patents at risk of being invalidated or interpreted narrowly, could place the patent applications at risk of not issuing, and/or could provoke third parties to assert claims against us or our current or future licensors, licensees or collaborators. We or our current or future licensors, licensees or collaborators may not prevail in any lawsuits that are initiated and the damages or other remedies awarded, if any, may not be commercially meaningful.
The duration of our intellectual property rights is limited.
Patents generally have a term of 20 years from the earliest effective filing date of the application that first discloses the claimed invention or an obvious variant thereof. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after the resulting products are commercialized. As a result, our owned and in-licensed patents may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours and we may need to rely solely on regulatory or similar protections, if they are available.
We expect to seek extensions of patent terms for our issued patents, where available. In the United States, this includes under the Hatch-Waxman Act, which permits a patent term extension of up to five years beyond the original expiration date of the patent as compensation for regulatory delays. However, such a patent term extension cannot lengthen the remaining term of a patent beyond a total of 14 years from the product’s approval date. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. During the period of patent term extension, the claims of the patent are not enforceable over their full scope, but instead are limited to the scope of the approved product. In addition, the applicable authorities, including the FDA in the United States, and any equivalent regulatory authority in other countries, may not agree with our assessment of whether such extensions are available and may refuse to grant extensions to our patents, or may grant more limited extensions than we request. In addition, we may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to the expiration of relevant patents or otherwise failing to satisfy applicable requirements. If this occurs, any period during which we have the right to exclusively market our product will be shorter than we would otherwise expect, and our competitors may obtain approval of and launch products earlier than might otherwise be the case.
Non-compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies could adversely affect our patent protection.
Periodic maintenance and annuity fees on issued United States patents and most foreign patent applications and patents must be paid to the U.S. Patent and Trademark Office, or USPTO, and
S-56
Table of Contents
foreign patent agencies, respectively, in order to maintain such patents and patent applications. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment, and other similar provisions during the patent application, examination and issuance processes. While an inadvertent lapse can be cured, in some cases, by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or our current or future licensors, licensees or collaborators fail to maintain the patents and patent applications covering our product candidates, our competitors might be able to enter the market with similar or identical products or technologies, which would have a material adverse effect on our business, financial condition and results of operations.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect our products and product candidates.
As is the case with other biotechnology and pharmaceutical companies, our success is heavily dependent on intellectual property rights, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves technological and legal complexity, and obtaining and enforcing biopharmaceutical patents is costly, time-consuming and inherently uncertain. The U.S. Supreme Court and the United States Court of Appeals for the Federal Circuit have ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances, weakening the rights of patent owners in certain situations or ruling that certain subject matter is not eligible for patent protection. In addition to increasing uncertainty with regard to our and our collaborator’s ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents once obtained. Depending on decisions by Congress, the federal courts, the USPTO and equivalent bodies in foreign jurisdictions, the laws and regulations governing patents could change in unpredictable ways that could weaken the ability of us and our current or future licensors, licensees or collaborators to obtain new patents or to enforce existing patents and patents we may obtain in the future.
We may be unable to obtain intellectual property rights or technologies necessary to develop and commercialize our product candidates.
Several third parties are actively researching and seeking and obtaining patent protection in the liver and metabolic diseases, retinal diseases and cancer fields, and there are issued third-party patents and published third-party patent applications in these fields. The patent landscape around our aldafermin, MK-3655 (NGM313), NGM120, NGM621, NGM707 and NGM438 product candidates is complex, and we are aware of several third-party patents and patent applications containing claims directed to compositions of matter, methods of use and related subject matter, some of which pertain, at least in part, to subject matter that might be relevant to our product candidates. However, we may not be aware of all third-party intellectual property rights potentially relating to our product candidates and technologies.
Depending on what patent claims ultimately issue and how courts construe the issued patent claims, as well as the ultimate formulation and method of use of our product candidates, we may need to obtain a license to practice the technology claimed in such patents. There can be no assurance that such licenses will be available on commercially reasonable terms, or at all. If a third party does not offer us a necessary license or offers a license only on terms that are unattractive or unacceptable to us, we might be unable to develop and commercialize one or more of our product candidates, which would have a material adverse effect on our business, financial condition and results of operations.
S-57
Table of Contents
Moreover, even if we obtain licenses to such intellectual property, but subsequently fail to meet our obligations under our license agreements, or such license agreements are terminated for any other reason, we may lose our rights to in-licensed technologies.
The licensing or acquisition of third-party intellectual property rights is a competitive area, and more established companies may pursue strategies to license or acquire third-party intellectual property rights that we may consider attractive or necessary. These established companies may have a competitive advantage over us due to their size, capital resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment, or at all. If we are unable to successfully obtain rights to required third-party intellectual property rights or maintain the existing intellectual property rights we have, we may have to abandon development of the relevant program or product candidate, which could have a material adverse effect on our business, financial condition, results of operations and prospects.
We could lose the ability to continue the development and commercialization of our products or product candidates if we breach any license agreement related to those products or product candidates.
Our commercial success depends upon our ability, and the ability of our current and future licensors, licensees and collaborators, to develop, manufacture, market and sell our products and product candidates and use our proprietary technologies without infringing the proprietary rights of third parties. A third party may hold intellectual property rights, including patent rights that are important or necessary to the development of our products. As a result, we are a party to a number of technology and patent licenses that are important to our business and we expect to enter into additional licenses in the future. If we fail to comply with the obligations under these agreements, including payment and diligence obligations, our licensors may have the right to terminate these agreements, in which event we may not be able to develop, manufacture, market or sell any product that is covered by these agreements or to engage in any other activities necessary to our business that require the freedom-to-operate afforded by the agreements, or we may face other penalties under the agreements. For example, we are party to license agreements with multiple vendors under which we license cell lines used to produce multiple product candidates, including some that are currently subject to our collaboration with Merck. We require prior consent from some of these vendors to grant sub-licenses under these agreements. Therefore, these vendors may be able to prevent us from granting sub-licenses to third parties, which could affect our ability or Merck’s ability to use certain desired manufacturers in order to manufacture our product candidates.
Any of the foregoing could materially adversely affect the value of the product or product candidate being developed under any such agreement. Termination of these agreements or reduction or elimination of our rights under these agreements may result in our having to negotiate new or amended agreements, which may not be available to us on equally favorable terms, or at all, or cause us to lose our rights under these agreements, including our rights to intellectual property or technology important to our development programs.
We may become involved in lawsuits or other proceedings to protect or enforce our intellectual property, which could be expensive, time-consuming and unsuccessful and have a material adverse effect on the success of our business.
Third parties may infringe patents or misappropriate or otherwise violate intellectual property rights owned or controlled by us or our current or future licensors, licensees or collaborators. In the future, it may be necessary to initiate legal proceedings to enforce or defend these intellectual property
S-58
Table of Contents
rights, to protect trade secrets or to determine the validity or scope of intellectual property rights that are owned or controlled by us or our current or future licensors, licensees or collaborators. Litigation could result in substantial costs and diversion of management resources, which could harm our business and financial results.
If we or our current or future licensors, licensees or collaborators initiated legal proceedings against a third party to enforce a patent covering a product candidate, the defendant could counterclaim that such patent is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. In an infringement or declaratory judgment proceeding, a court may decide that a patent owned by or licensed to us or our current or future licensors, licensees or collaborators is invalid or unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that the patent does not cover the technology in question. An adverse result in any litigation proceeding could put one or more of the patents at risk of being invalidated, narrowed, held unenforceable or interpreted in such a manner that would not preclude third parties from entering the market with competing products.
Third parties may initiate legal proceedings against us or our current or future licensors, licensees or collaborators to challenge the validity or scope of intellectual property rights we own or control. For example, generic or biosimilar drug manufacturers or other competitors or third parties may challenge the scope, validity or enforceability of patents owned or controlled by us or our current or future licensors, licensees or collaborators. These proceedings can be expensive, lengthy and time-consuming, and many of our adversaries may have the ability to dedicate substantially greater resources to prosecuting these legal actions than us. Accordingly, despite our efforts, we or our current or future licensors, licensees or collaborators may not be able to prevent third parties from infringing upon or misappropriating intellectual property rights we own, control or have rights to, particularly in countries where the laws may not protect those rights as fully as in the United States.
There is a risk that some of our confidential information could be compromised by disclosure during litigation because of the substantial amount of discovery required. Additionally, many foreign jurisdictions have rules of discovery that are different than those in the United States and that may make defending or enforcing our patents extremely difficult. There also could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of shares of our common stock.
Third-party pre-issuance submission of prior art to the USPTO, opposition, derivation, revocation, reexamination, inter partes review or interference proceedings, or other pre-issuance or post-grant proceedings, as well as other patent office proceedings or litigation in the United States or other jurisdictions brought by third parties against patents or patent applications owned or controlled by us or our current or future licensors, licensees or collaborators, may be necessary to determine the inventorship, priority, patentability or validity of these patents or patent applications. An unfavorable outcome could leave our technology or product candidates without patent protection and allow third parties to commercialize our technology or product candidates without payment to us. Additionally, potential licensees or collaborators could be dissuaded from collaborating with us to license, develop or commercialize current or future product candidates if the breadth or strength of protection provided by our patents and patent applications is threatened. Even if we successfully defend such litigation or proceeding, we may incur substantial costs and it may distract our management and other employees.
S-59
Table of Contents
Third parties may initiate legal proceedings against us alleging that we infringe their intellectual property rights or we may initiate legal proceedings against third parties to challenge the validity or scope of the third-party intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability and that of our current or future licensors, licensees or collaborators to develop, manufacture, market and sell any product candidates that we may develop and use our proprietary technologies without infringing, misappropriating or otherwise violating the intellectual property and proprietary rights of third parties. Third parties may initiate legal proceedings against us or our current or future licensors, licensees or collaborators alleging that we infringe their intellectual property rights. Alternatively, we may initiate legal proceedings to challenge the validity or scope of intellectual property rights controlled by third parties, including in oppositions, interferences, revocations, reexaminations, inter partes review or derivation proceedings before the USPTO or its counterparts in other jurisdictions. These proceedings can be expensive, lengthy and time-consuming, and many of our adversaries may have the ability to dedicate substantially greater resources to prosecuting these legal actions than us.
Even if we believe third-party intellectual property claims are without merit, there is no assurance that a court would find in our favor on questions of infringement, validity, enforceability or priority. In order to successfully challenge the validity of any such U.S. patent in federal court, we would need to overcome a presumption of validity in favor of the granted third party patent. This is a high burden, requiring us to present clear and convincing evidence as to the invalidity of any such U.S. patent claim.
An unfavorable outcome in any such proceeding could require us and our current or future licensors, licensees or collaborators to cease using the related technology or developing or commercializing the product or product candidate, or to attempt to license rights to it from the prevailing party, which may not be available on commercially reasonable terms, or at all. Additionally, we could be found liable for monetary damages, including treble damages and attorneys’ fees, if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business.
We perform searches of patent and scientific databases in order to identify documents that may be of potential relevance to the freedom-to-operate of our product candidates. Such searches generally are conducted based on keywords, amino acid and nucleic acid sequences, inventors/authors and assignees/entities to capture U.S. and European patents and patent applications, PCT publications and scientific journal articles. There can be no assurance that these searches will identify all potentially relevant patents or patent applications, and the failure to identify any such patents or patent applications could have a material adverse effect on the commercialization of our product candidates.
We are aware of several third-party patents and patent applications containing claims directed to compositions of matter, methods of use and related subject matter, some of which pertain, at least in part, to subject matter that might be relevant to our aldafermin, MK-3655 (NGM313), NGM120, NGM621 NGM707 and NGM438 product candidates. We are not aware of any facts that would lead us to conclude that the valid and enforceable claims of any third-party patents would reasonably be interpreted to encompass our product candidates, unless we are unsuccessful in our opposition of any of the granted European patents that are discussed below, or any appeals stemming therefrom. As to pending third-party applications, we cannot predict with any certainty the claims that will issue, if any, or the scope of such issued claims.
We filed an opposition in the European Patent Office, or EPO, against a patent, granted to St. Vincent’s Hospital Sydney Limited, or St. Vincent’s, claiming the use of MIC-1, also known as GDF15, for the treatment of obesity. The Opposition Division of the EPO upheld the patent as granted in the
S-60
Table of Contents
first instance proceedings. We appealed this decision to the Board of Appeals but subsequently withdrew from the opposition. The opposition proceedings were terminated with the patent being maintained. If we were to decide to continue development of NGM395, we may not be able to commercially launch NGM395 in Europe for the treatment of obesity without a license from St. Vincent’s or until after the patent’s expiration, which is currently scheduled for April 2025. There is no assurance that such a license, if necessary, can be obtained from St. Vincent’s on commercially reasonable terms, or at all.
We filed an opposition in the EPO against a patent, granted to Amgen, claiming the use of GDF15 polypeptides for the treatment of several metabolic disorders, including obesity. At the first instance proceedings, the Opposition Division of the EPO maintained the patent in view of amendments to the patent and statements by Amgen disclaiming the treatment of obesity. We appealed to the Board of Appeals at the EPO the Opposition Division’s decision to maintain the Amgen patent. If we were to decide to continue development of NGM395 and should the patent be upheld on appeal or should we decide not to pursue the appeal, we may not be able to commercially launch NGM395 in Europe for the treatment of any metabolic orders encompassed by the claims without a license from Amgen or until after the patent’s expiration, which is currently scheduled for April 2032. There is no assurance that such a license, if necessary, can be obtained from Amgen on commercially reasonable terms, or at all.
Our inability to protect our confidential information and trade secrets would harm our business and competitive position.
In addition to seeking patents for some of our technology and products, we also rely substantially on trade secrets in our activities, including unpatented know-how, technology and other proprietary materials and information, to maintain our competitive position. We seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants. However, these steps may be inadequate, we may fail to enter into agreements with all such parties or any of these parties may breach the agreements and disclose our proprietary information and there may be no adequate remedy available for such breach of an agreement. We cannot assure that our proprietary information will not be disclosed or that we can meaningfully protect our trade secrets. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts both within and outside the United States may be less willing, or unwilling, to protect trade secrets. If a competitor lawfully obtained or independently developed any of our trade secrets, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position.
Third parties may initiate legal proceedings against us alleging that we or our employees have misappropriated their intellectual property or claiming ownership of what we regard as our own intellectual property.
Many of our employees, including our senior management, were previously employed at universities or at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Most of these employees executed proprietary rights, non-disclosure and non-competition agreements in connection with such previous employment. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees have used or disclosed confidential information or intellectual property, including trade secrets or other proprietary information, of any such employee’s former employer, or that third parties have an interest in our patents as an inventor or co-inventor. Litigation may be necessary to defend against these claims. If we fail in prosecuting or defending any
S-61
Table of Contents
such claims, we may lose valuable intellectual property rights or personnel, pay monetary damages and/or sustain other damages. Such intellectual property rights could be awarded to a third party, and we could be required to obtain a license from such third party to commercialize our technology or products. Such a license may not be available on commercially reasonable terms, or at all. Even if we successfully prosecute or defend against such claims, litigation could result in substantial costs and distract management.
We may be subject to claims challenging the inventorship of our patents and other intellectual property or otherwise be unable to secure ownership of our intellectual property.
We and our current or future licensors, licensees or collaborators may be subject to claims that former employees, collaborators or other third parties have an interest in our owned or in-licensed patents, trade secrets, or other intellectual property as an inventor or co-inventor. Litigation may be necessary to defend against these and other claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, intellectual property that is important to our products and product candidates. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
It is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us. However, we may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property that we regard as our own. The assignment of intellectual property rights may not be self-executing, or the assignment agreements may be breached, and we may be forced to bring claims against third parties, or defend claims that they may bring against us, to determine the ownership of what we regard as our intellectual property. Such claims could have a material adverse effect on our business, financial condition, results of operations and prospects.
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
| • | others may be able to make products that are similar to any product candidates we may develop or utilize similar technologies that are not covered by the claims of the patents that we license or may own in the future; |
| • | we, or our current or future licensors, licensees or collaborators, might not have been the first to make the inventions covered by the issued patents and pending patent applications that we license or may own in the future; |
| • | we, or our current or future licensors, licensees or collaborators, might not have been the first to file patent applications covering certain of our or their inventions; |
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our owned or licensed intellectual property rights; |
| • | it is possible that our pending patent applications or those that we may own in the future will not lead to issued patents; |
| • | issued patents to which we hold rights may be held invalid or unenforceable, including as a result of legal challenges by our competitors; |
S-62
Table of Contents
| • | our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| • | we may not develop additional proprietary technologies that are patentable; |
| • | the patents of others may harm our business; and |
| • | we may choose not to file a patent application in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property. |
Should any of these events occur, they could have a material adverse effect on our business, financial condition, results of operations and prospects.
Risks Related to this Offering and Ownership of Our Common Stock
The market price of our common stock has been and may continue to be volatile, and you could lose all or part of your investment.
The market price for our common stock has fluctuated significantly from time to time, for example, varying between a high of $32.05 on December 30, 2020 and a low of $8.81 on October 7, 2019. The trading price of our common stock has been and may continue to be highly volatile and subject to wide fluctuations in response to various factors, some of which we cannot control. In addition to the factors discussed in this “Risk Factors” section, these factors include:
| • | developments associated with our collaboration with Merck, including Merck’s failure to exercise its remaining unilateral option to extend the research phase of the collaboration, any termination of the collaboration or other change in our relationship with Merck; |
| • | the success of competitive products or technologies, including disclosure of interim data by our competitors; |
| • | regulatory actions with respect to our product candidates or our competitors’ product candidates or products; |
| • | results of clinical trials of our product candidates or those of our competitors; |
| • | actual or anticipated changes in our growth rate relative to our competitors; |
| • | announcements by us or our competitors or collaborators of significant acquisitions, strategic collaborations, joint ventures, collaborations or capital commitments; |
| • | regulatory, legal or payor developments in the United States and other countries; |
| • | developments or disputes concerning patent applications, issued patents or other proprietary rights; |
| • | the recruitment or departure of key personnel; |
| • | the level of expenses related to any of our product candidates or clinical development programs; |
| • | the results of our efforts to in-license or acquire additional product candidates or products; |
| • | actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts; |
| • | variations in our financial results or those of companies that are perceived to be similar to us; |
| • | fluctuations in the valuation of companies perceived by investors to be comparable to us; |
S-63
Table of Contents
| • | share price and volume fluctuations attributable to inconsistent trading volume levels of our shares; |
| • | announcement or expectation of additional financing efforts; |
| • | sales of our common stock by us, our insiders or our other stockholders; |
| • | changes in the structure of healthcare payment systems; |
| • | market conditions in the pharmaceutical and biotechnology sectors; and |
| • | general economic, industry and market conditions. |
In addition, the stock market in general, and The Nasdaq Global Select Market and biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies, including in connection with the ongoing COVID-19 pandemic, which has resulted in decreased stock prices for many companies notwithstanding the lack of a fundamental change in their underlying business models or prospects. Broad market and industry factors, including worsening economic conditions and other adverse effects or developments relating to the evolving effects of the COVID-19 pandemic, may negatively affect the market price of our common stock, regardless of our actual operating performance. The realization of any of the above risks or any of a broad range of other risks, including those described in this “Risk Factors” section, could have a dramatic and material adverse impact on the market price of our common stock.
Because of potential volatility in our trading price and trading volume, we may incur significant costs from class action securities litigation.
Holders of stock in companies that have a volatile stock price frequently bring securities class action litigation against the company that issued the stock. We may be the target of this type of litigation in the future. If any of our stockholders were to bring a lawsuit of this type against us, even if the lawsuit is without merit, we could incur substantial costs defending the lawsuit and the time and attention of our management could be diverted from other business concerns, either of which could seriously harm our business. Refer also to the risk factor entitled “Sales of a substantial number of shares of our common stock in the public market could cause our stock price to fall.”
An active trading market for our common stock may not be sustained.
Our common stock is currently listed on The Nasdaq Global Select Market under the symbol “NGM” and trades on that market. We cannot ensure that an active trading market for our common stock will be sustained. Accordingly, we cannot ensure the liquidity of any trading market, your ability to sell your shares of our common stock when desired or the prices that you may obtain for your shares.
We will have broad discretion in the use of proceeds from this offering and may invest or spend the proceeds in ways with which you do not agree and in ways that may not yield a return.
We will have broad discretion over the use of proceeds from this offering. You may not agree with our decisions, and our use of the proceeds may not yield any return on your investment in us. Our failure to apply the net proceeds of this offering effectively could result in financial losses that could materially impair our ability to pursue our growth strategy, cause the price of our common stock to decline, delay development of our product candidates or require us to raise additional capital.
Our principal stockholders, including entities affiliated with The Column Group, Merck and management, own a substantial percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
Our executive officers, directors, significant stockholders, including entities affiliated with The Column Group and Merck, and their respective affiliates beneficially own a substantial amount of our
S-64
Table of Contents
voting stock. These stockholders may be able to determine all matters requiring stockholder approval. For example, these stockholders may be able to control elections of directors, amendments of our organizational documents or approval of any merger, sale of assets or other major corporate transaction. The interests of this group of stockholders may not always coincide with your interests or the interests of other stockholders. In addition, if any of our significant stockholders decide to sell a meaningful amount of their ownership position and there is not sufficient demand in the market for such stocks, our stock price could fall.
We are an “emerging growth company” as defined in the JOBS Act and will be able to avail ourselves of reduced disclosure requirements applicable to emerging growth companies, which could make our common stock less attractive to investors and adversely affect the market price of our common stock.
For so long as we remain an “emerging growth company” as defined in the JOBS Act, we may take advantage of certain exemptions from various requirements applicable to public companies that are not “emerging growth companies” including:
| • | the provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that our independent registered public accounting firm provide an attestation report on the effectiveness of our internal control over financial reporting; |
| • | the “say on pay” provisions (requiring a non-binding stockholder vote to approve compensation of certain executive officers) and the “say on golden parachute” provisions (requiring a non-binding stockholder vote to approve golden parachute arrangements for certain executive officers in connection with mergers and certain other business combinations) of the Dodd-Frank Wall Street Reform and Protection Act, or the Dodd-Frank Act, and some of the disclosure requirements of the Dodd-Frank Act relating to compensation of our chief executive officer; and |
| • | the requirement to provide detailed compensation discussion and analysis in proxy statements and reports filed under the Exchange Act, and instead provide a reduced level of disclosure concerning executive compensation. |
Because our independent registered public accounting firm is not required to provide an attestation report on the effectiveness of our internal control over financial reporting so long as we qualify as an “emerging growth company,” the risk that material weaknesses or significant deficiencies in our internal control over financial reporting go undetected may be increased. Likewise, our election not to provide certain information, including certain financial information and certain information regarding compensation of our executive officers, that we would otherwise have been required to provide in filings we make with the SEC, may make it more difficult for investors and securities analysts to evaluate our company.
We may take advantage of these reporting exemptions until we are no longer an “emerging growth company”, which in certain circumstances could be for up to five years. We will cease to be an “emerging growth company” upon the earliest of: (1) December 31, 2024; (2) the last day of the first fiscal year in which our annual gross revenue is $1.07 billion or more; (3) the date on which we have, during the previous rolling three-year period, issued more than $1 billion in non-convertible debt securities; and (4) the date on which we are deemed to be a “large accelerated filer” as defined in the Exchange Act.
With respect to the JOBS Act, we are also taking advantage of some, but not all, of the reduced regulatory and reporting requirements that are available to us so long as we qualify as an “emerging growth company.” For example, we are not subject to the same new or revised accounting standards
S-65
Table of Contents
as other public companies that are not “emerging growth companies.” As a result, changes in U.S. generally accepted accounting principles or their interpretation, the adoption of new guidance or the application of existing guidance to changes in our business could significantly affect our financial position and results of operations.
We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions and reduced requirements. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock, and our stock price may be more volatile and may decline. In addition, if we lose our “emerging growth company” status sooner than anticipated, we may incur additional costs to comply with rules and regulations required for public companies, which may impact our financial position and results of operations.
Sales of a substantial number of shares of our common stock in the public market could cause our stock price to fall.
For the trading days during the three months ended September 30, 2020, the average daily trading volume for our common stock on The Nasdaq Global Select Market was only 180,030 shares. As a result, sales of a substantial number of shares of our common stock in the public market, including pursuant to the Sales Agreement or the perception in the market that we or the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock. In addition, as a result of the low trading volume of our common stock, the trading of relatively small quantities of shares by our stockholders could disproportionately influence the market price of our common stock in either direction. The price for our shares could, for example, decline significantly in the event that a large number of shares of our common stock are sold on the market without commensurate demand, as compared to an issuer with a higher trading volume that could better absorb those sales without an adverse impact on its stock price. Moreover, certain holders of our common stock have rights, subject to certain conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders. Shares issued to Merck in the private placement that occurred concurrently with our IPO became available for sale in the public market beginning on March 17, 2020, subject to the conditions of Rule 144 under the Securities Act of 1933, as amended, or the Securities Act.
If you purchase shares of common stock in this offering, you will suffer immediate dilution of your investment.
The price of our common stock in this offering is higher than the net tangible book value per share of our common stock. Therefore, if you purchase shares of our common stock in this offering, you will pay a price per share that substantially exceeds our net tangible book value per share after this offering. To the extent outstanding options are exercised, you will incur further dilution. Our net tangible book value as of September 30, 2020 was $276.1 million, or $4.01 per share. As a result, if you purchase shares of our common stock in this offering, you will incur an immediate dilution of $21.66 in net tangible book value per share from the price you paid. See the section of this prospectus supplement entitled “Dilution” for a more detailed discussion of the dilution you will incur if you purchase common stock in this offering.
If we issue additional shares of common stock, or securities convertible into or exchangeable or exercisable for shares of common stock, including pursuant to the Sales Agreement, our stockholders, including investors who purchase shares of common stock in this offering, will experience additional dilution, and any such issuances may result in downward pressure on the price of our common stock. We also cannot assure you that we will be able to sell shares or other securities in any other offering at a price per share that is equal to or greater than the price per share paid by investors in this offering, and investors purchasing shares or other securities in the future could have rights superior to existing stockholders.
S-66
Table of Contents
We do not intend to pay dividends on our common stock so any returns will be limited to the value of our stock.
We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to investors in this offering will therefore be limited to the appreciation of their stock, if any.
Some provisions of our charter documents, Delaware law and our agreement with Merck may have anti-takeover effects or could otherwise discourage an acquisition of us by others, even if an acquisition would benefit our stockholders, and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and amended and restated bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us or increase the cost of acquiring us, even if doing so would benefit our stockholders, or to remove our current management. These provisions include:
| • | a board of directors divided into three classes serving staggered three-year terms, such that not all members of the board will be elected at one time; |
| • | authorizing the issuance of “blank check” preferred stock, the terms of which we may establish and shares of which we may issue without stockholder approval; |
| • | prohibiting cumulative voting in the election of directors, which would otherwise allow for less than a majority of stockholders to elect director candidates; |
| • | prohibiting stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders; |
| • | eliminating the ability of stockholders to call a special meeting of stockholders; and |
| • | establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings. |
These provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, who are responsible for appointing the members of our management. Because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, or DGCL, which may discourage, delay or prevent someone from acquiring us or merging with us whether or not it is desired by or beneficial to our stockholders. In addition, Section 203 of the DGCL prohibits a publicly-held Delaware corporation from engaging in a business combination with an interested stockholder, which is generally a person that together with its affiliates owns, or within the last three years has owned, 15% of our voting stock, for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner.
Certain provisions in our agreement with Merck may also deter a change of control. For example, under our agreement with Merck, a change of control gives Merck the right to terminate the research phase of the collaboration as well as additional rights if our acquirer is a qualifying large pharmaceutical company or has a research, development or commercialization program that competes with a program licensed by Merck.
S-67
Table of Contents
Any provision of our amended and restated certificate of incorporation, amended and restated bylaws, Delaware law or our agreement with Merck that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock, and could also affect the price that some investors are willing to pay for our common stock.
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware and the federal district courts of the United States will be the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware is the exclusive forum for the following types of actions or proceedings under Delaware statutory or common law: any derivative action or proceeding brought on our behalf; any action asserting a breach of a fiduciary duty; any action asserting a claim against us arising pursuant to the DGCL, our amended and restated certificate of incorporation or our amended and restated bylaws; or any action asserting a claim against us that is governed by the internal affairs doctrine. This provision would not apply to suits brought to enforce a duty or liability created by the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. Furthermore, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all such Securities Act actions. Accordingly, both state and federal courts have jurisdiction to entertain such claims. To prevent having to litigate claims in multiple jurisdictions and the threat of inconsistent or contrary rulings by different courts, among other considerations, our amended and restated certificate of incorporation further provides that the federal district courts of the United States of America will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act. While the Delaware courts have determined that such choice of forum provisions are facially valid, a stockholder may nevertheless seek to bring a claim in a venue other than those designated in the exclusive forum provisions. In such instance, we would expect to vigorously assert the validity and enforceability of the exclusive forum provisions of our amended and restated certificate of incorporation. This may require significant additional costs associated with resolving such action in other jurisdictions and there can be no assurance that the provisions will be enforced by a court in those other jurisdictions.
These exclusive forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage lawsuits against us and our directors, officers and other employees. If a court were to find either exclusive-forum provision in our amended and restated certificate of incorporation to be inapplicable or unenforceable in an action, we may incur further significant additional costs associated with resolving the dispute in other jurisdictions, all of which could seriously harm our business.
General Risk Factors
New tax laws or regulations, changes to existing tax laws or regulations or changes in their application to us or our customers may have a material adverse effect on our business, cash flows, financial condition or results of operations.
New income, sales, use or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could adversely affect our business and financial condition. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, the 2017 Tax Act sanctioned many significant changes to the U.S. tax laws. Future guidance from the U.S. Internal Revenue Service, or IRS, and other tax
S-68
Table of Contents
authorities with respect to the 2017 Tax Act may affect us, and certain aspects of the 2017 Tax Act may be repealed or modified in future legislation. For example, the CARES Act modified certain provisions of the 2017 Tax Act. Changes in corporate tax rates, the realization of net deferred tax assets relating to our operations, the taxation of foreign earnings and the deductibility of expenses under the 2017 Tax Act or future reform legislation could have a material impact on the value of our deferred tax assets, could result in significant one-time charges and could increase our future U.S. tax expense.
Future changes in financial accounting standards or practices may cause adverse and unexpected revenue fluctuations and adversely affect our reported results of operations.
Future changes in financial accounting standards may cause adverse, unexpected revenue fluctuations and affect our reported financial position or results of operations. Financial accounting standards in the United States are constantly under review and new pronouncements and varying interpretations of pronouncements have occurred frequently in the past and are expected to occur again in the future. As a result, we may be required to make changes in our accounting policies. Those changes could affect our financial condition and results of operations or the way in which such financial condition and results of operations are reported. Compliance with new accounting standards may also result in additional expenses. As a result, we intend to invest all reasonably necessary resources to comply with evolving standards, and this investment may result in increased general and administrative expenses and a diversion of management time and attention from business activities to compliance activities.
We incur increased costs as a result of operating as a public company, and our management devotes substantial time to new compliance initiatives.
As a public company, we incur significant legal, accounting, insurance and other expenses that we did not incur as a private company, and these expenses may increase even more after we are no longer an “emerging growth company.” As a public company, we are subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act and the Dodd-Frank Act, as well as rules adopted, and to be adopted, by the SEC and The Nasdaq Global Select Market. Our management and other personnel devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations increase our legal and financial compliance costs and may make some activities more time-consuming and costly. The increased costs will increase our net loss. For example, these rules and regulations make it more difficult and more expensive for us to obtain director and officer liability insurance and we may be required to incur substantial costs to maintain sufficient coverage. We cannot predict or estimate the amount or timing of additional costs we may incur in the future to respond to these requirements. The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as executive officers.
Specifically, in order to comply with the requirements of being a public company, we need to undertake various actions, including implementing new internal controls and procedures and hiring new accounting or internal audit staff. The Sarbanes-Oxley Act requires that we maintain effective disclosure controls and procedures and internal control over financial reporting. We are continuing to develop and refine our disclosure controls and other procedures that are designed to ensure that information required to be disclosed by us in the reports that we file with the SEC is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that information required to be disclosed in reports under the Exchange Act is accumulated and communicated to our principal executive and financial officers. Any failure to develop or maintain effective controls could adversely affect the results of periodic management evaluations. In the event that we are not able to demonstrate compliance with the Sarbanes-Oxley Act, that our internal control
S-69
Table of Contents
over financial reporting is perceived as inadequate or that we are unable to produce timely or accurate financial statements, investors may lose confidence in our operating results and the price of our common stock could decline. In addition, if we are unable to continue to meet these requirements, we may not be able to remain listed on The Nasdaq Global Select Market.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We designed our disclosure controls and procedures to reasonably assure that information we must disclose in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well-conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected.
If securities or industry analysts do not publish research, or publish inaccurate or unfavorable research, about our business, our stock price and trading volume could decline.
Our stock price and trading volume is heavily influenced by the way analysts and investors interpret our clinical trial results, financial information and other disclosures. If securities or industry analysts do not publish research or reports about our business, delay publishing reports about our business or publish negative reports about our business, regardless of accuracy, our stock price and trading volume could decline. The trading market for our common stock depends, in part, on the research and reports that securities or industry analysts publish about us or our business. We do not have any control over these analysts. A limited number of analysts are currently covering our company. If the number of analysts that cover us declines, demand for our common stock could decrease and our common stock price and trading volume may decline. Even if our common stock is actively covered by analysts, we do not have any control over the analysts or the measures that analysts or investors may rely upon to forecast our future results. Over-reliance by analysts or investors on any particular metric to forecast our future results may result in forecasts that differ significantly from our own.
S-70
Table of Contents
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus supplement, the accompanying prospectus and the documents we have filed with the SEC that are incorporated by reference contain “forward-looking statements” within the meaning of Section 27A of the Securities Act, and Section 21E of the Exchange Act. These statements relate to future events or to our future operating or financial performance and involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. Forward-looking statements may include, but are not limited to, statements about:
| • | the success, cost and timing of our product development activities and clinical trials, and the initiation of, enrollment in, availability of data for and other events related to such trials; |
| • | our or our partners’ ability to obtain and maintain regulatory approval for aldafermin (NGM282), MK-3655 (NGM313), NGM621, NGM120, NGM707, NGM438 and any of our future product candidates, and any related restrictions, limitations and/or warnings in the label of an approved product candidate; |
| • | our belief that aldafermin may have the potential to be a treatment for NASH patients with moderate to advanced fibrosis; |
| • | the potential roles of ILT2, ILT4 and LAIR1 in cancer and the potential consequences of ILT2, ILT4 and LAIR1 blockade; |
| • | our belief regarding the impact of our product candidate side effects and our ability to effectively manage these side effects; |
| • | our belief that MK-3655 (NGM313) may have the potential to be a treatment for NASH patients with early to moderate fibrosis; |
| • | the potential renewal of our research collaboration, product development and license agreement with Merck and the possibility that Merck will decide to exercise its option to license certain programs upon our completion of a proof-of-concept study in humans; |
| • | our ability to obtain funding for our operations; |
| • | the commercialization of our product candidates, if approved; |
| • | our plans to research, develop and commercialize our product candidates; |
| • | our ability to attract additional collaborators with development, regulatory and commercialization expertise; |
| • | current and future agreements with third parties in connection with the potential commercialization of aldafermin, MK-3655 (NGM313), NGM621, NGM120, NGM707, NGM438 or any other future approved product; |
| • | the size and growth potential of the markets for our product candidates, and our ability to serve those markets; |
| • | the rate and degree of market acceptance of our product candidates, as well as the reimbursement coverage for our product candidates; |
| • | regulatory developments in the United States and foreign countries; |
| • | the performance of, and our ability to obtain sufficient supply of clinical trial material in a timely manner from, third-party suppliers and manufacturers; |
| • | the success of competing therapies that are or may become available; |
| • | our ability to attract and retain key scientific, development and management personnel; |
S-71
Table of Contents
| • | our estimates regarding future expenses, revenue, capital requirements and needs for additional financing; |
| • | our expectations regarding the period during which we qualify as an emerging growth company under the JOBS Act; |
| • | our expectations regarding our ability to obtain, maintain, protect and enforce intellectual property protection for our product candidates; and |
| • | our anticipated use of the net proceeds from this offering. |
In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,” “would,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “projects,” “predicts,” “potential” and similar expressions intended to identify forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and are subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. We discuss in greater detail many of these risks under the heading “Risk Factors” contained in this prospectus supplement. Also, these forward-looking statements represent our estimates and assumptions only as of the date of the document containing the applicable statement. Unless required by law, we undertake no obligation to update or revise any forward-looking statements to reflect new information or future events or developments. Thus, you should not assume that our silence over time means that actual events are bearing out as expressed or implied in such forward-looking statements. You should read this prospectus supplement and the accompanying prospectus, together with the documents we have filed with the SEC that are incorporated by reference and any free writing prospectus that we may authorize for use in connection with this offering completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of the forward-looking statements in the foregoing documents by these cautionary statements.
S-72
Table of Contents
We estimate that the net proceeds from this offering will be approximately $117.0 million, or approximately $134.7 million if the underwriters exercise in full their option to purchase additional shares of common stock, after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
We intend to use the net proceeds from this offering for working capital and other general corporate purposes, which may include funding our pipeline of development programs, general and administrative activities and capital expenditures. Pending the application of the net proceeds, we expect to invest the proceeds in investment-grade, interest-bearing instruments or other securities. We will retain broad discretion over the use of the net proceeds from this offering.
S-73
Table of Contents
Dilution is the amount by which the price paid by the purchasers of the shares of common stock sold in the offering exceeds the net tangible book value per share of common stock after the offering. Net tangible book value per share is determined by subtracting our total liabilities from the total book value of our tangible assets and dividing the difference by the number of shares of common stock deemed to be outstanding at that date.
Our historical net tangible book value as of September 30, 2020 was $276.1 million, or $4.01 per share.
After giving effect to the sale of 4,629,630 shares of common stock in this offering at the public offering price of $27.00 per share, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us, our as adjusted net tangible book value as of September 30, 2020 would have been approximately $393.1 million, or $5.34 per share. This represents an immediate increase in as adjusted net tangible book value of $1.33 per share to our existing stockholders and immediate dilution of $21.66 per share to new investors purchasing common stock in this offering.
The following table illustrates this dilution on a per share basis to new investors participating in this offering:
| Public offering price per share |
$ | 27.00 | ||||||
|
|
|
|||||||
| Historical net tangible book value per share as of September 30, 2020 |
$ | 4.01 | ||||||
| Increase per share attributable to new investors participating in this offering |
1.33 | |||||||
|
|
|
|||||||
| As adjusted net tangible book value per share after giving effect to this offering |
5.34 | |||||||
|
|
|
|||||||
| Dilution in adjusted net tangible book value per share to new investors participating in this offering |
$ | 21.66 | ||||||
|
|
|
If the underwriters exercise in full their option to purchase additional shares of our common stock at the public offering price of $27.00 per share, the as adjusted net tangible book value per share after giving effect to this offering would be $5.53 per share, representing an immediate increase to existing stockholders of $1.52 per share, and immediate dilution to new investors participating in this offering of $21.47 per share.
The foregoing discussion and table are based on 68,934,767 shares of our common stock outstanding as of September 30, 2020, and exclude, in each case as of September 30, 2020, the following:
| • | 10,651,475 shares of our common stock issuable upon the exercise of outstanding stock options with a weighted-average exercise price of $10.03 per share; |
| • | 6,309,573 shares of our common stock available for issuance or future grant under the EIP; |
| • | 788,120 shares of our common stock available for issuance under the ESPP; and |
| • | 21,930 shares of our common stock reserved for future issuance under the Matching Plan. |
The foregoing discussion and table do not reflect any automatic (or other) increases in the number of shares of common stock reserved for future issuance under the EIP, the ESPP or the Matching Plan, including the 2,823,565 shares of common stock automatically added to the EIP on January 1, 2021 in accordance with the terms of the EIP. In addition, the foregoing discussion and
S-74
Table of Contents
table also do not include up to $150.0 million of our common stock that remained available for sale at September 30, 2020 under the Sales Agreement. Between September 30, 2020 and the date of this prospectus supplement, 809,700 shares were sold under the Sales Agreement at an average price of $27.94 per share for net proceeds of $21.9 million, after deducting sales commissions. As of the date of this prospectus supplement, $127.4 million of common stock remained available to be sold under the Sales Agreement, subject to certain conditions as specified in the agreement.
To the extent that any of the outstanding options are exercised there could be further dilution to new investors participating in this offering. In addition, we may choose to raise additional capital, including pursuant to the Sales Agreement, due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. To the extent that we raise additional capital through the sale of equity securities, the issuance of such securities could result in further dilution to our stockholders.
S-75
Table of Contents
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings to support our operations and finance the growth and development of our business. We do not intend to pay cash dividends on our common stock for the foreseeable future. Any future determination related to our dividend policy will be made at the discretion of our board of directors and will depend upon, among other factors, our results of operations, financial condition, capital requirements, contractual restrictions, business prospects and other factors our board of directors may deem relevant.
S-76
Table of Contents
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO NON-U.S. HOLDERS
The following is a discussion of the material U.S. federal income tax consequences applicable to non-U.S. holders (as defined below) with respect to their purchase, ownership and disposition of shares of our common stock issued pursuant to this offering, but does not purport to be a complete analysis of all potential tax effects. All prospective non-U.S. holders of our common stock should consult their tax advisors with respect to the U.S. federal income tax consequences of the purchase, ownership and disposition of our common stock, as well as any consequences arising under the laws of any other taxing jurisdiction, including any state, local and non-U.S. tax consequences and any U.S. federal non-income tax consequences. In general, a non-U.S. holder means a beneficial owner of our common stock (other than a partnership or an entity or arrangement treated as a partnership for U.S. federal income tax purposes) that is not a U.S. holder. A U.S. holder is, for U.S. federal income tax purposes:
| • | an individual who is a citizen or resident of the United States; |
| • | a corporation, or an entity treated as a corporation for U.S. federal income tax purposes, created or organized in the United States or under the laws of the United States or of any state thereof or the District of Columbia; |
| • | an estate, the income of which is subject to U.S. federal income tax regardless of its source; or |
| • | a trust if (1) a U.S. court can exercise primary supervision over the trust’s administration and one or more U.S. persons have the authority to control all of the trust’s substantial decisions or (2) the trust has a valid election in effect under applicable U.S. Treasury Regulations to be treated as a U.S. person. |
This discussion is based on current provisions of the Code, existing U.S. Treasury Regulations promulgated thereunder, published administrative rulings and judicial decisions, all as in effect as of the date of this prospectus supplement. These laws are subject to change and to differing interpretation, possibly with retroactive effect. Any change or differing interpretation could alter the tax consequences to non-U.S. holders described in this prospectus supplement.
This discussion is limited to non-U.S. holders that purchase our common stock pursuant to this offering and hold shares of our common stock as a capital asset within the meaning of Section 1221 of the Code (generally, for investment). This discussion does not address all aspects of U.S. federal income taxation that may be relevant to a particular non-U.S. holder in light of that non-U.S. holder’s individual circumstances, nor does it address certain special tax rules applicable to particular non-U.S. holders, such as holders that own, or are deemed to own, more than 5% of our capital stock (except to the extent specifically set forth below), corporations that accumulate earnings to avoid U.S. federal income tax, tax-exempt organizations, banks, financial institutions, insurance companies, brokers, dealers or traders in securities, commodities or currencies, tax-qualified retirement plans, holders who hold or receive our common stock pursuant to the exercise of employee stock options or otherwise as compensation, holders holding our common stock as part of a hedge, straddle or other risk reduction strategy, conversion transaction or other integrated investment, holders deemed to sell our common stock under the constructive sale provisions of the Code, “qualified foreign pension funds” as defined in Section 897(l)(2) of the Code and entities all of the interests of which are held by qualified foreign pension funds, controlled foreign corporations, passive foreign investment companies and certain former citizens or long-term residents of the United States. This discussion also does not address the effect of the alternative minimum tax or the Medicare contribution tax on net investment income, the impact of special tax accounting rules under Section 451(b) of the Code, or any aspects of U.S. estate or gift tax.
In addition, this discussion does not address the tax treatment of partnerships (or entities or arrangements that are treated as partnerships for U.S. federal income tax purposes) or persons that
S-77
Table of Contents
hold our common stock through such partnerships, entities or arrangements. If a partnership, including any entity or arrangement treated as a partnership for U.S. federal income tax purposes, holds shares of our common stock, the U.S. federal income tax treatment of a partner in such partnership will generally depend upon the status of the partner, the activities of the partnership and certain determinations made at the partner level. Such partners and partnerships should consult their tax advisors regarding the tax consequences of the purchase, ownership and disposition of our common stock.
There can be no assurance that the IRS will not challenge one or more of the tax consequences described herein, and we have not obtained, nor do we intend to obtain, a ruling from the IRS regarding the U.S. federal income tax consequences with respect to the matters discussed below.
Distributions on our Common Stock
As described in the section entitled “dividend policy,” we do not anticipate declaring or paying dividends to holders of our common stock in the foreseeable future. However, distributions, if any, on our common stock generally will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. If a distribution exceeds our current and accumulated earnings and profits, the excess will be treated as a tax-free return of the non-U.S. holder’s investment, up to such holder’s adjusted tax basis in the common stock. Any remaining excess will be treated as capital gain from the sale or exchange of such common stock, subject to the tax treatment described below in “Gain on Sale, Exchange or Other Disposition of Our Common Stock.”
Subject to the discussions below regarding effectively connected income, backup withholding and foreign accounts, dividends paid to a non-U.S. holder generally will be subject to withholding of U.S. federal income tax at a 30% rate, or such lower rate as may be specified by an applicable income tax treaty. A non-U.S. holder that is eligible for a reduced rate of U.S. withholding tax under an income tax treaty may obtain a refund or credit of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS. A non-U.S. holder of our common stock who claims the benefit of an applicable income tax treaty generally will be required to provide a properly executed IRS Form W-8BEN or W-8BEN-E (or successor form) and satisfy relevant certification and other requirements. Non-U.S. holders are urged to consult their tax advisors regarding their entitlement to benefits under a relevant income tax treaty.
Dividends that are treated as effectively connected with a trade or business conducted by a non-U.S. holder within the United States and, if an applicable income tax treaty so provides, that are attributable to a permanent establishment or a fixed base maintained by the non-U.S. holder within the United States, generally are exempt from the 30% withholding tax if the non-U.S. holder satisfies applicable certification requirements. To claim the exemption, the non-U.S. holder must furnish to us or the applicable withholding agent a valid IRS Form W-8ECI (or applicable successor form), certifying that the dividends are effectively connected with the non-U.S. holder’s conduct of a trade or business within the United States. However, such U.S. effectively connected income, net of specified deductions and credits, is taxed at the U.S. federal income tax rates applicable to a “United States person” (as defined in the Code), unless a specific treaty exemption applies. Any U.S. effectively connected income received by a non-U.S. holder that is a corporation may also, under certain circumstances, be subject to an additional “branch profits tax” at a 30% rate or such lower rate as may be specified by an applicable income tax treaty.
Non-U.S. holders that do not provide the required certification on a timely basis, but qualify for a reduced treaty rate, may obtain a refund of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS. Non U.S. holders should consult their tax advisors regarding any applicable income tax treaties that may provide for different rules.
S-78
Table of Contents
Gain on Sale, Exchange or Other Disposition of our Common Stock
Subject to the discussions below regarding backup withholding and foreign accounts, in general, a non-U.S. holder will not be subject to any U.S. federal income tax on any gain realized upon such holder’s sale, exchange or other disposition of shares of our common stock unless:
| • | the gain is effectively connected with a U.S. trade or business of the non-U.S. holder and, if an applicable income tax treaty so provides, is attributable to a permanent establishment or a fixed base maintained in the United States by such non-U.S. holder, in which case the non-U.S. holder generally will be taxed at the U.S. federal income tax rates applicable to United States persons and, if the non-U.S. holder is a foreign corporation, the branch profits tax described above in “Distributions on our Common Stock” also may apply; |
| • | the non-U.S. holder is a nonresident alien individual who is present in the United States for 183 days or more in the taxable year of the disposition and certain other conditions are met, in which case the non-U.S. holder will be subject to a 30% tax (or such lower rate as may be specified by an applicable income tax treaty) on the net gain derived from the disposition, which may be offset by certain U.S. source capital losses of the non-U.S. holder, if any (even though the individual is not considered a resident of the United States), provided the non-U.S. holder has timely filed U.S. federal income tax returns with respect to such losses; or |
| • | our common stock constitutes a U.S. real property interest because we are, or have been, at any time during the five-year period preceding such disposition (or the non-U.S. holder’s holding period, if shorter) a “United States real property holding corporation.” Even if we are or become a United States real property holding corporation, provided that our common stock is “regularly traded” (as defined by U.S. Treasury Regulations) on an established securities market, our common stock will be treated as a U.S. real property interest only with respect to a non-U.S. holder that holds more than 5% of our outstanding common stock, directly or indirectly, actually or constructively, during the shorter of the five-year period ending on the date of the disposition or the period that the non-U.S. holder held our common stock. In such case, such non-U.S. holder generally will be taxed on its net gain derived from the disposition at the U.S. federal income tax rates applicable to United States persons. Generally, a corporation is a United States real property holding corporation only if the fair market value of its U.S. real property interests equals or exceeds 50% of the sum of (i) the fair market value of its worldwide real property interests and (ii) its other assets used or held for use in a trade or business. Although there can be no assurance, we do not believe that we are, or have been, a United States real property holding corporation, or that we are likely to become one in the future. No assurance can be provided that our common stock will be regularly traded on an established securities market for purposes of the rules described above. |
Backup Withholding and Information Reporting
We must report annually to the IRS and to each non-U.S. holder the gross amount of distributions on our common stock paid to such holder, regardless of whether such distributions constitute dividends or whether any tax was withheld. A non-U.S. holder will have to comply with specific certification procedures to establish that it is not a United States person in order to avoid backup withholding at the applicable rate with respect to dividends on our common stock. U.S. backup withholding generally will not apply to a non-U.S. holder who provides a properly executed IRS Form W-8BEN or W-8BEN-E (or successor form) or otherwise establishes an exemption.
Information reporting and backup withholding generally will apply to the proceeds of a disposition of our common stock by a non-U.S. holder effected by or through the U.S. office of any broker, U.S. or foreign, unless the holder certifies its status as a non-U.S. holder and satisfies certain other requirements, or otherwise establishes an exemption. Generally, information reporting and backup
S-79
Table of Contents
withholding will not apply to a payment of disposition proceeds to a non-U.S. holder where the transaction is effected outside the United States through a non-U.S. office of a broker. However, for information reporting purposes, dispositions effected through a non-U.S. office of a broker with substantial U.S. ownership or operations generally will be treated in a manner similar to dispositions effected through a U.S. office of a broker.
Non-U.S. holders should consult their tax advisors regarding the application of the information reporting and backup withholding rules to them.
Copies of information returns may be made available to the tax authorities of the country in which the non-U.S. holder resides or is incorporated under the provisions of a specific treaty or agreement.
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules from a payment to a non-U.S. holder may be allowed as a credit against the non-U.S. holder’s U.S. federal income tax liability, if any, and may entitle such holder to a refund, provided that the required information is timely furnished to the IRS.
Foreign Accounts
Sections 1471 through 1474 of the Code, or commonly referred to as FATCA, generally impose a U.S. federal withholding tax of 30% on certain payments made to a “foreign financial institution” (as specifically defined for this purpose), unless such institution enters into an agreement with the U.S. government to, among other things, withhold on certain payments and collect and provide to the U.S. tax authorities substantial information regarding U.S. account holders of such institution (which may include certain equity and debt holders of such institution, as well as certain account holders that are foreign entities with U.S. owners). Foreign financial institutions located in jurisdictions that have an intergovernmental agreement with the United States governing these withholding and reporting requirements may be subject to different rules. FATCA also generally imposes a 30% withholding tax on certain payments made to a non-financial foreign entity, unless such entity provides the withholding agent with either a certification that it does not have any substantial direct or indirect U.S. owners, or information regarding substantial direct and indirect U.S. owners of the entity. The withholding tax under FATCA described above will not apply if the foreign financial institution or non-financial foreign entity otherwise qualifies for an exemption from the rules. The FATCA withholding provisions described above currently apply to dividends on our common stock. The FATCA withholding provisions also would apply to the gross proceeds of a disposition of our common stock, except that the U.S. Treasury Department has released proposed regulations which, if finalized in their present form, would eliminate such withholding. In its preamble to such proposed regulations, the U.S. Treasury Department stated that taxpayers generally may rely on the proposed regulations until final regulations are issued.
Under certain circumstances, a non-U.S. holder might be eligible for refunds or credits of such taxes. Non-U.S. holders are encouraged to consult with their tax advisors regarding the possible implications of FATCA on their investment in our common stock.
EACH PROSPECTIVE INVESTOR SHOULD CONSULT ITS TAX ADVISORS REGARDING THE TAX CONSEQUENCES OF PURCHASING, HOLDING AND DISPOSING OF OUR COMMON STOCK, AS WELL AS TAX CONSEQUENCES ARISING UNDER ANY STATE, LOCAL, NON-U.S. OR U.S. FEDERAL NON-INCOME TAX LAWS.
S-80
Table of Contents
We and the underwriters named below have entered into an underwriting agreement with respect to the shares being offered. Subject to certain conditions, each underwriter has severally agreed to purchase the number of shares indicated in the following table. Goldman Sachs & Co. LLC, Citigroup Global Markets Inc. and Cowen and Company, LLC are the representatives of the underwriters.
| Underwriters |
Number of Shares |
|||
| Goldman Sachs & Co. LLC |
1,990,741 | |||
| Citigroup Global Markets Inc. |
1,203,704 | |||
| Cowen and Company, LLC |
925,926 | |||
| Raymond James & Associates, Inc. |
277,778 | |||
| B. Riley Securities, Inc. |
231,481 | |||
|
|
|
|||
| Total |
4,629,630 | |||
|
|
|
|||
The underwriters are committed to take and pay for all of the shares being offered, if any are taken, other than the shares covered by the option described below unless and until this option is exercised.
The underwriters have an option to buy up to an additional 694,444 shares of common stock from us. They may exercise that option for 30 days. If any shares are purchased pursuant to this option, the underwriters will severally purchase shares in approximately the same proportion as set forth in the table above.
The following table shows the per share and total underwriting discounts and commissions to be paid to the underwriters by us. Such amounts are shown assuming both no exercise and full exercise of the underwriters’ option to purchase up to 694,444 additional shares.
| Paid by Us |
No Exercise | Full Exercise | ||||||
| Per Share |
$ | 1.62 | $ | 1.62 | ||||
| Total |
$ | 7,500,001 | $ | 8,625,000 | ||||
Shares sold by the underwriters to the public will initially be offered at the public offering price set forth on the cover of this prospectus. Any shares sold by the underwriters to securities dealers may be sold at a discount of up to $0.972 per share from the public offering price. After the initial offering of the shares, the representatives may change the offering price and the other selling terms. The offering of the shares by the underwriters is subject to receipt and acceptance and subject to the underwriters’ right to reject any order in whole or in part.
We, our executive officers and our directors have agreed with the underwriters, subject to certain exceptions, not to dispose of or hedge any of our common stock or securities convertible into or exchangeable for shares of our common stock during the period from the date of this prospectus supplement continuing through (i) the date 90 days after the date of this prospectus supplement with respect to us, and (ii) the date 60 days after the date of this prospectus supplement with respect to our executive officers and directors, except with the prior written consent of the representatives. This agreement does not apply to any existing employee benefit plans.
The restrictions in the immediately preceding paragraph do not apply to our executive officers or directors in certain circumstances, including (i) the transfers not for value of our common stock as bona fide gifts, by will, to an immediate family member or to certain trusts; (ii) to the extent the party to the lock-up agreement is an entity, the transfer of our common stock to affiliates, limited partners, general partners, limited liability company members or stockholders; (iii) transfers of our common stock pursuant to a bona fide third-party tender offer, merger, consolidation or other similar transaction made
S-81
Table of Contents
to all of our stockholders and involving a change of control of us; (iv) in connection with a sale of shares of our common stock acquired in open market transactions after the date of this prospectus and, other than for our directors and officers, shares of our common stock purchased in this offering, whether or not issuer-directed; (v) transfers of our common stock to us for the net exercise of options granted pursuant to our equity incentive plans described elsewhere in this prospectus or to cover tax withholding for grants pursuant to our equity incentive plans or in connection with the repurchase of shares of our common stock issued pursuant to an employee benefit plan described elsewhere in this prospectus; and (vi) transfers of our common stock pursuant to existing 10b5-1 trading plans under the Exchange Act. The exceptions described in (i) through (iii) above are subject to a requirement that the transferee enter into a lockup agreement with the underwriters containing similar restrictions, the exceptions described in (i) and (ii) above are subject to a requirement that no public announcement or filing under Section 16 of the Exchange Act shall be required or voluntarily made during the restricted period and any such transfer shall not involve a disposition for value, the exceptions described in (iv) and (v) above are subject to a requirement that no public announcement or filing under Section 16 of the Exchange Act shall be required or voluntarily made during the restricted period and the exception described in (vi) above is subject to a requirement that any public reports or filings including filings under Sections 13 or 16 of the Exchange Act that shall be required to be made or voluntarily made shall clearly indicate in the footnotes that such sale was made pursuant to a trading plan established pursuant to Rule 10b5-1 under the Exchange Act. The restrictions in the immediately preceding paragraph do not apply to us in certain circumstances, including (i) the sale of shares in this offering, (ii) the issuance of shares upon the exercise of an option or warrant or conversion of a security outstanding on the date of this prospectus supplement, (iii) the issuance of securities pursuant to our equity incentive plans, (iv) the filing of any registration statement (including amendments or prospectuses thereto) (A) on Form S-8 or any successor form thereto with respect to the registration of securities to be offered under an employee benefit or equity incentive plans or (B) that we may be required to file pursuant to that certain Amended and Restated Investor Rights Agreement, dated as of March 20, 2015, by and between us and the investor parties thereto, (v) the issuance of shares of common stock to Merck and/or its designate affilate(s) and the entry into a purchase agreement related thereto, in each case pursuant to the exercise by Merck of its option to acquire shares of common stock from us pursuant to that certain Letter Agreement, dated as of March 20, 2015, by and between Merck and us, or (vi) the issuance of securities in connection with transactions that include a commercial relationship (including without limitation, joint ventures, marketing or distribution arrangements, collaboration agreements or intellectual property license agreements) or any acquisition by us of any of our subsidiaries of the securities, business, property or other assets of another person or entity pursuant to any employee benefit plan assumed by us in connection with such acquisition, and the issuance of any such securities pursuant to such agreement; provided further, that, in the case of clause (vi), the aggregate number of shares that we may sell or issue or agree to sell or issue pursuant to clause (vi) shall not exceed 5% of the total number of shares of common stock issued and outstanding immediately following the completion of this offering, and provided further that we shall cause each recipient of such securities pursuant to clause (vi) to execute and deliver to the representatives a lock-up agreements described above.
The representatives, in their sole discretion, may release the common stock and other securities subject to the lock-up agreements described above in whole or in part at any time.
Our common stock is listed on The Nasdaq Global Select Market under the symbol “NGM.”
In connection with the offering, the underwriters may purchase and sell shares of our common stock in the open market. These transactions may include short sales, stabilizing transactions and purchases to cover positions created by short sales. Short sales involve the sale by the underwriters of a greater number of shares than they are required to purchase in the offering, and a short position represents the amount of such sales that have not been covered by subsequent purchases. A “covered
S-82
Table of Contents
short position” is a short position that is not greater than the amount of additional shares for which the underwriters’ option described above may be exercised. The underwriters may cover any covered short position by either exercising their option to purchase additional shares or purchasing shares in the open market. In determining the source of shares to cover the covered short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase additional shares pursuant to the option described above. “Naked” short sales are any short sales that create a short position greater than the amount of additional shares for which the option described above may be exercised. The underwriters must cover any such naked short position by purchasing shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there may be downward pressure on the price of our common stock in the open market after pricing that could adversely affect investors who purchase in the offering. Stabilizing transactions consist of various bids for or purchases of our common stock made by the underwriters in the open market prior to the completion of the offering.
The underwriters may also impose a penalty bid. This occurs when a particular underwriter repays to the underwriters a portion of the underwriting discount received by it because the representatives have repurchased shares sold by or for the account of such underwriter in stabilizing or short covering transactions.
Purchases to cover a short position and stabilizing transactions, as well as other purchases by the underwriters for their own accounts, may have the effect of preventing or retarding a decline in the market price of our stock, and together with the imposition of the penalty bid, may stabilize, maintain or otherwise affect the market price of our common stock. As a result, the price of the common stock may be higher than the price that otherwise might exist in the open market. The underwriters are not required to engage in these activities and may end any of these activities at any time. These transactions may be effected on The Nasdaq Global Select Market, in the over-the-counter market or otherwise.
We estimate that our total expenses of the offering, excluding underwriting discounts and commissions, will be approximately $465,000. We have agreed to reimburse the underwriters for expenses of up to $15,000 related to the clearance of this offering with the Financial Industry Regulatory Authority, Inc. and compliance with state securities or “blue sky” laws.
We have agreed to indemnify the several underwriters against certain liabilities, including liabilities under the Securities Act.
The underwriters and their respective affiliates are full service financial institutions engaged in various activities, which may include sales and trading, commercial and investment banking, advisory, investment management, investment research, principal investment, hedging, market making, brokerage and other financial and non-financial activities and services. Certain of the underwriters and their respective affiliates have provided, and may in the future provide, a variety of these services to us and to persons and entities with relationships with us, for which they received or will receive customary fees and expenses.
In the ordinary course of their various business activities, the underwriters and their respective affiliates, officers, directors and employees may purchase, sell or hold a broad array of investments and actively trade securities, derivatives, loans, commodities, currencies, credit default swaps and other financial instruments for their own account and for the accounts of their customers, and such investment and trading activities may involve or relate to our assets, securities and/or instruments (directly, as collateral securing other obligations or otherwise) and/or persons and entities with relationships with us. The underwriters and their respective affiliates may also communicate independent investment recommendations, market color or trading ideas and/or publish or express
S-83
Table of Contents
independent research views in respect of such assets, securities or instruments and may at any time hold, or recommend to clients that they should acquire, long and/or short positions in such assets, securities and instruments.
Notice to Prospective Investors in the European Economic Area and the United Kingdom
In relation to each Member State of the European Economic Area and the United Kingdom (each a “Relevant State”), no common shares (the “Shares”) have been offered or will be offered pursuant to the offering to the public in that Relevant State prior to the publication of a prospectus in relation to the Shares which has been approved by the competent authority in that Relevant State or, where appropriate, approved in another Relevant State and notified to the competent authority in that Relevant State, all in accordance with the Prospectus Regulation, except that offers of Shares may be made to the public in that Relevant State at any time under the following exemptions under the Prospectus Regulation:
| (a) | to any legal entity which is a qualified investor as defined under the Prospectus Regulation; |
| (b) | to fewer than 150 natural or legal persons (other than qualified investors as defined under the Prospectus Regulation), subject to obtaining the prior consent of representatives for any such offer; or |
| (c) | in any other circumstances falling within Article 1(4) of the Prospectus Regulation, |
provided that no such offer of Shares shall require us or any underwriter to publish a prospectus pursuant to Article 3 of the Prospectus Regulation or supplement a prospectus pursuant to Article 23 of the Prospectus Regulation.
For the purposes of this provision, the expression an “offer to the public” in relation to any Shares in any Relevant State means the communication in any form and by any means of sufficient information on the terms of the offer and any Shares to be offered so as to enable an investor to decide to purchase or subscribe for any Shares, and the expression “Prospectus Regulation” means Regulation (EU) 2017/1129.
The above selling restriction is in addition to any other selling restrictions set out below.
Notice to Prospective Investors in the United Kingdom
This prospectus supplement and the accompanying prospectus are only being distributed to, and are only directed at, persons in the United Kingdom that are qualified investors within the meaning of Article 2(1)(e) of the Prospectus Directive that are also (i) investment professionals falling within Article 19(5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005 (the “Order”) or (ii) high net worth entities, and other persons to whom it may lawfully be communicated, falling within Article 49(2)(a) to (d) of the Order (each such person being referred to as a “relevant person”). This prospectus supplement and its contents are confidential and should not be distributed, published or reproduced (in whole or in part) or disclosed by recipients to any other persons in the United Kingdom. Any person in the United Kingdom that is not a relevant person should not act or rely on this document or any of its contents.
Notice to Prospective Investors in Canada
The securities may be sold in Canada only to purchasers purchasing, or deemed to be purchasing, as principal that are accredited investors, as defined in National
S-84
Table of Contents
Instrument 45-106 Prospectus Exemptions or subsection 73.3(1) of the Securities Act (Ontario), and are permitted clients, as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant Obligations. Any resale of the securities must be made in accordance with an exemption from, or in a transaction not subject to, the prospectus requirements of applicable securities laws.
Securities legislation in certain provinces or territories of Canada may provide a purchaser with remedies for rescission or damages if this prospectus supplement (including any amendment thereto) contains a misrepresentation, provided that the remedies for rescission or damages are exercised by the purchaser within the time limit prescribed by the securities legislation of the purchaser’s province or territory. The purchaser should refer to any applicable provisions of the securities legislation of the purchaser’s province or territory for particulars of these rights or consult with a legal advisor.
Pursuant to section 3A.3 of National Instrument 33-105 Underwriting Conflicts (NI 33-105), the underwriters are not required to comply with the disclosure requirements of NI 33-105 regarding underwriter conflicts of interest in connection with this offering.
Notice to Prospective Investors in Hong Kong
The shares may not be offered or sold in Hong Kong by means of any document other than (i) in circumstances which do not constitute an offer to the public within the meaning of the Companies (Winding Up and Miscellaneous Provisions) Ordinance (Cap. 32 of the Laws of Hong Kong) (“Companies (Winding Up and Miscellaneous Provisions) Ordinance”) or which do not constitute an invitation to the public within the meaning of the Securities and Futures Ordinance (Cap. 571 of the Laws of Hong Kong) (“Securities and Futures Ordinance”), or (ii) to “professional investors” as defined in the Securities and Futures Ordinance and any rules made thereunder, or (iii) in other circumstances which do not result in the document being a “prospectus” as defined in the Companies (Winding Up and Miscellaneous Provisions) Ordinance, and no advertisement, invitation or document relating to the shares may be issued or may be in the possession of any person for the purpose of issue (in each case whether in Hong Kong or elsewhere), which is directed at, or the contents of which are likely to be accessed or read by, the public in Hong Kong (except if permitted to do so under the securities laws of Hong Kong) other than with respect to shares which are or are intended to be disposed of only to persons outside Hong Kong or only to “professional investors” in Hong Kong as defined in the Securities and Futures Ordinance and any rules made thereunder.
Notice to Prospective Investors in Singapore
This prospectus supplement has not been registered as a prospectus with the Monetary Authority of Singapore. Accordingly, this prospectus supplement and any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of the shares may not be circulated or distributed, nor may the shares be offered or sold, or be made the subject of an invitation for subscription or purchase, whether directly or indirectly, to persons in Singapore other than (i) to an institutional investor (as defined under Section 4A of the Securities and Futures Act, Chapter 289 of Singapore (the “SFA”)) under Section 274 of the SFA, (ii) to a relevant person (as defined in Section 275(2) of the SFA) pursuant to Section 275(1) of the SFA, or any person pursuant to Section 275(1A) of the SFA, and in accordance with the conditions specified in Section 275 of the SFA or (iii) otherwise pursuant to, and in accordance with the conditions of, any other applicable provision of the SFA, in each case subject to conditions set forth in the SFA.
Where the shares are subscribed or purchased under Section 275 of the SFA by a relevant person which is a corporation (which is not an accredited investor (as defined in Section 4A of the SFA)) the sole business of which is to hold investments and the entire share capital of which is owned by one or more individuals, each of whom is an accredited investor, the securities (as defined in Section 239(1) of the SFA) of that corporation shall not be transferable for 6 months after that
S-85
Table of Contents
corporation has acquired the shares under Section 275 of the SFA except: (1) to an institutional investor under Section 274 of the SFA or to a relevant person (as defined in Section 275(2) of the SFA), (2) where such transfer arises from an offer in that corporation’s securities pursuant to Section 275(1A) of the SFA, (3) where no consideration is or will be given for the transfer, (4) where the transfer is by operation of law, (5) as specified in Section 276(7) of the SFA, or (6) as specified in Regulation 32 of the Securities and Futures (Offers of Investments) (Shares and Debentures) Regulations 2005 of Singapore (“Regulation 32”).
Where the shares are subscribed or purchased under Section 275 of the SFA by a relevant person which is a trust (where the trustee is not an accredited investor (as defined in Section 4A of the SFA)) whose sole purpose is to hold investments and each beneficiary of the trust is an accredited investor, the beneficiaries’ rights and interest (howsoever described) in that trust shall not be transferable for 6 months after that trust has acquired the shares under Section 275 of the SFA except: (1) to an institutional investor under Section 274 of the SFA or to a relevant person (as defined in Section 275(2) of the SFA), (2) where such transfer arises from an offer that is made on terms that such rights or interest are acquired at a consideration of not less than S$200,000 (or its equivalent in a foreign currency) for each transaction (whether such amount is to be paid for in cash or by exchange of securities or other assets), (3) where no consideration is or will be given for the transfer, (4) where the transfer is by operation of law, (5) as specified in Section 276(7) of the SFA, or (6) as specified in Regulation 32.
Notice to Prospective Investors in Japan
The securities have not been and will not be registered under the Financial Instruments and Exchange Act of Japan (Act No. 25 of 1948, as amended), or the FIEA. The securities may not be offered or sold, directly or indirectly, in Japan or to or for the benefit of any resident of Japan (including any person resident in Japan or any corporation or other entity organized under the laws of Japan) or to others for reoffering or resale, directly or indirectly, in Japan or to or for the benefit of any resident of Japan, except pursuant to an exemption from the registration requirements of the FIEA and otherwise in compliance with any relevant laws and regulations of Japan.
Notice to Prospective Investors in Australia
No prospectus or other disclosure document, as defined in the Corporations Act 2001 (“Cth”) of Australia, or the Corporations Act, in relation to our shares has been or will be lodged with the Australian Securities & Investments Commission ( the “ASIC”). This document has not been lodged with ASIC and is only directed to certain categories of exempt persons. Accordingly, if you receive this document in Australia:
(a) you confirm and warrant that you are either:
(i) a “sophisticated investor” under section 708(8)(a) or (b) of the Corporations Act;
(ii) a “sophisticated investor” under section 708(8)(c) or (d) of the Corporations Act and that you have provided an accountant’s certificate to us which complies with the requirements of section 708(8)(c)(i) or (ii) of the Corporations Act and related regulations before the offer has been made;
(iii) a person associated with us under section 708(12) of the Corporations Act; or
(iv) a “professional investor” within the meaning of section 708(11)(a) or (b) of the Corporations Act, and to the extent that you are unable to confirm or warrant that you are an exempt sophisticated investor, associated person or professional investor under the Corporations Act, any offer made to you under this document is void and incapable of acceptance; and
S-86
Table of Contents
(b) you warrant and agree that you will not offer any of our shares for resale in Australia within 12 months of that security being issued unless any such resale offer is exempt from the requirement to issue a disclosure document under section 708 of the Corporations Act.
Notice to Prospective Investors in Israel
In the State of Israel this prospectus supplement shall not be regarded as an offer to the public to purchase shares of our common stock under the Israeli Securities Law, 5728 – 1968, or the Israeli Securities Law, which requires a prospectus to be published and authorized by the Israel Securities Authority if it complies with certain provisions of Section 15 of the Israeli Securities Law, including, inter alia, if: (i) the offer is made, distributed or directed to not more than 35 investors, subject to certain conditions (the “Addressed Investors”); or (ii) the offer is made, distributed or directed to certain qualified investors defined in the First Addendum of the Israeli Securities Law, subject to certain conditions (the “Qualified Investors”). The Qualified Investors shall not be taken into account in the count of the Addressed Investors and may be offered to purchase securities in addition to the 35 Addressed Investors. We have not and will not take any action that would require it to publish a prospectus in accordance with and subject to the Israeli Securities Law. We have not and will not distribute this prospectus supplement or make, distribute or direct an offer to subscribe for our common stock to any person within the State of Israel, other than to Qualified Investors and up to 35 Addressed Investors.
Qualified Investors may have to submit written evidence that they meet the definitions set out in of the First Addendum to the Israeli Securities Law. In particular, we may request, as a condition to be offered shares of our common stock, that Qualified Investors will each represent, warrant and certify to us and/or to anyone acting on our behalf: (i) that it is an investor falling within one of the categories listed in the First Addendum to the Israeli Securities Law; (ii) which of the categories listed in the First Addendum to the Israeli Securities Law regarding Qualified Investors is applicable to it; (iii) that it will abide by all provisions set forth in the Israeli Securities Law and the regulations promulgated thereunder in connection with the offer to be issued shares of our common stock; (iv) that the shares of our common stock and that it will be issued are subject to exemptions available under the Israeli Securities Law (a) for its own account, (b) for investment purposes only and (c) not issued with a view to resale within the State of Israel, other than in accordance with the provisions of the Israeli Securities Law; and (v) that it is willing to provide further evidence of its Qualified Investor status. Addressed Investors may have to submit written evidence in respect of their identity and may have to sign and submit a declaration containing, inter alia, the Addressed Investor’s name, address and passport number or Israeli identification number.
S-87
Table of Contents
Cooley LLP is serving as our counsel in this offering. Davis Polk & Wardwell LLP of Menlo Park, California is representing the underwriters in connection with this offering. GC&H Investments, LLC, an entity that includes current and former partners and associates of Cooley LLP, beneficially owns 10,000 shares of our common stock.
Ernst & Young LLP, independent registered public accounting firm, has audited our consolidated financial statements included in our Annual Report on Form 10-K for the year ended December 31, 2019, as set forth in their report (which contains an explanatory paragraph describing a change in accounting principle as described in Note 2 to the consolidated financial statements), which is incorporated by reference in this prospectus supplement and elsewhere in the registration statement. Our consolidated financial statements are incorporated by reference in reliance on Ernst & Young LLP’s report, given on their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
This prospectus supplement and the accompanying prospectus are part of the registration statement on Form S-3 that we filed with the SEC. The registration statement that contains this prospectus supplement, including the exhibits and schedules to the registration statement, contains additional information about us and the securities offered by this prospectus supplement and the accompanying prospectus. For further information with respect to us and the securities we are offering under this prospectus supplement and the accompanying prospectus, we refer you to the registration statement and the exhibits and schedules filed as a part of the registration statement. You should rely only on information contained in this prospectus supplement, the accompanying prospectus or incorporated by reference into this prospectus supplement and the accompanying prospectus. We have not authorized any person to provide you with different information. We are not making an offer of these securities in any state where the offer is not permitted. You should not assume that the information in this prospectus supplement is accurate as of any date other than the date on the front page of this prospectus supplement, regardless of the time of delivery of this prospectus supplement or any sale of the securities offered by this prospectus supplement. We file annual, quarterly and current reports, proxy statements and other information with the SEC. Our SEC filings are available to the public at the SEC’s website at http://www.sec.gov.
We maintain a website at www.ngmbio.com. Information contained in or accessible through our website does not constitute a part of this prospectus supplement or the accompanying prospectus and is not incorporated by reference into this prospectus supplement or the accompanying prospectus.
S-88
Table of Contents
INCORPORATION OF CERTAIN INFORMATION BY REFERENCE
The SEC allows us to incorporate by reference the information we file with it, which means that we can disclose important information to you by referring you to another document that we have filed separately with the SEC. You should read the information incorporated by reference because it is an important part of this prospectus supplement and the accompanying prospectus. We incorporate by reference the following information or documents that we have filed with the SEC (Commission File No. 000-38853):
| • | our Annual Report on Form 10-K for the year ended December 31, 2019, filed with the SEC on March 17, 2020; |
| • | our Definitive Proxy Statement on Schedule 14A, filed with the SEC on April 8, 2020 (excluding those portions that are not incorporated by reference into our Annual Report on Form 10-K for the year ended December 31, 2019); |
| • | our Quarterly Reports on Form 10-Q for the quarter ended March 31, 2020, filed with the SEC on May 13, 2020, for the quarter ended June 30, 2020, filed with the SEC on August 12, 2020 and for the quarter ended September 30, 2020, filed with the SEC on November 12, 2020; |
| • | our Current Reports on Form 8-K filed with the SEC on February 13, 2020, February 24, 2020, March 9, 2020, May 27, 2020 and June 8, 2020; and |
| • | the description of our common stock set forth in our registration statement on Form 8-A, filed with the SEC on March 29, 2019, including any amendment or report filed for the purpose of updating such description, including Exhibit 4.3 of our Annual Report on Form 10-K for the year ended December 31, 2019. |
We also incorporate by reference any future filings (other than current reports furnished under Item 2.02 or Item 7.01 of Form 8-K and exhibits filed on such form that are related to such items) made with the SEC pursuant to Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act, until we file a post-effective amendment which indicates the termination of the offering of the securities made by this prospectus supplement and the accompanying prospectus. Information in such future filings updates and supplements the information provided in this prospectus supplement and the accompanying prospectus. Any statements in any such future filings will automatically be deemed to modify and supersede any information in any document we previously filed with the SEC that is incorporated or deemed to be incorporated herein by reference to the extent that statements in the later filed document modify or replace such earlier statements. We will provide to each person, including any beneficial owner, to whom a prospectus supplement or the underlying prospectus is delivered, without charge upon the written or oral request, a copy of any or all of the documents that are incorporated by reference in this prospectus supplement but not delivered with the prospectus, including exhibits that are specifically incorporated by reference in such documents. You should direct any requests for documents to:
NGM Biopharmaceuticals, Inc.
333 Oyster Point Boulevard
South San Francisco, California 94080
(650) 392-1768
Attn: Secretary
S-89
Table of Contents
Prospectus

$400,000,000
Common Stock
Preferred Stock
Debt Securities
Warrants
From time to time, we may offer and sell up to an aggregate amount of $400,000,000 of any combination of the securities described in this prospectus, either individually or in combination with other securities. We may also offer common stock or preferred stock upon conversion of debt securities, common stock upon conversion of preferred stock, or common stock, preferred stock or debt securities upon the exercise of warrants.
We will provide the specific terms of these offerings and securities in one or more supplements to this prospectus. We may also authorize one or more free writing prospectuses to be provided to you in connection with these offerings. The prospectus supplement and any related free writing prospectus may also add, update or change information contained in this prospectus. You should carefully read this prospectus, the applicable prospectus supplement and any related free writing prospectus, as well as any documents incorporated by reference into this prospectus, before buying any of the securities being offered.
Our common stock is listed on The Nasdaq Global Select Market under the symbol “NGM.” On June 2, 2020, the last reported sale price of our common stock was $20.37 per share. The applicable prospectus supplement will contain information, where applicable, as to other listings, if any, on The Nasdaq Global Select Market or other securities exchange of the securities covered by the applicable prospectus supplement.
Investing in our securities involves a high degree of risk. Before making an investment decision, you should review carefully the risks described under the heading “Risk Factors” on page 5 of this prospectus and any similar section contained in the applicable prospectus supplement and in any free writing prospectuses we have authorized for use in connection with a specific offering, and under similar headings in the documents that are incorporated by reference into this prospectus.
This prospectus may not be used to consummate a sale of securities unless accompanied by a prospectus supplement.
The securities may be sold directly by us to investors, through agents designated from time to time or to or through underwriters or dealers, on a continuous or delayed basis. The supplements to this prospectus will provide the specific terms of the plan of distribution. If any agents or underwriters are involved in the sale of any securities with respect to which this prospectus is being delivered, the names of such agents or underwriters and any applicable fees, commissions, discounts and options to purchase additional shares will be set forth in a prospectus supplement. The price to the public of such securities and the net proceeds we expect to receive from such sale will also be set forth in a prospectus supplement.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is June 16, 2020.
Table of Contents
| 1 | ||||
| 5 | ||||
| 6 | ||||
| 8 | ||||
| 9 | ||||
| 14 | ||||
| 20 | ||||
| 22 | ||||
| 25 | ||||
| 27 | ||||
| 27 | ||||
| 27 | ||||
| 27 | ||||
Table of Contents
ABOUT THIS PROSPECTUS
This prospectus is part of a registration statement on Form S-3 that we filed with the Securities and Exchange Commission, or SEC, using a “shelf” registration process. Under this shelf registration statement, we may, from time to time, offer and sell, either individually or in combination, in one or more offerings, up to a total dollar amount of $400,000,000 of any combination of the securities described in this prospectus.
This prospectus provides you with a general description of the securities we may offer. Each time we offer securities under this prospectus, we will provide a prospectus supplement that will contain more specific information about the terms of that offering. We may also authorize one or more free writing prospectuses to be provided to you that may contain material information relating to these offerings. The prospectus supplement and any related free writing prospectus that we may authorize to be provided to you may also add, update or change any of the information contained in this prospectus or in the documents that we have incorporated by reference into this prospectus. We urge you to read carefully this prospectus, any applicable prospectus supplement and any free writing prospectuses we have authorized for use in connection with a specific offering, together with the information incorporated herein by reference as described under the heading “Incorporation of Certain Information by Reference,” before buying any of the securities being offered.
This prospectus may not be used to consummate a sale of securities unless it is accompanied by a prospectus supplement.
We have not authorized anyone to provide you with any information or to make any representations other than those contained in, or incorporated by reference into, this prospectus, any applicable prospectus supplement or any free writing prospectuses prepared by or on behalf of us or to which we have referred you. We take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. This prospectus, any applicable supplement to this prospectus or any related free writing prospectus do not constitute an offer to sell or the solicitation of an offer to buy any securities other than the registered securities to which they relate, nor do this prospectus, any applicable supplement to this prospectus or any related free writing prospectus constitute an offer to sell or the solicitation of an offer to buy securities in any jurisdiction to any person to whom it is unlawful to make such offer or solicitation in such jurisdiction. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of the common stock and the distribution of this prospectus outside the United States.
The information appearing in this prospectus, any applicable prospectus supplement or any related free writing prospectus is accurate only as of the date on the front of the document and any information we have incorporated by reference is accurate only as of the date of the document incorporated by reference, regardless of the time of delivery of this prospectus, the applicable prospectus supplement or any related free writing prospectus, or any sale of a security. Our business, financial condition, results of operations and prospects may have changed since those dates.
This prospectus contains and incorporates by reference market data and industry statistics and forecasts that are based on independent industry publications and other publicly available information. Although we believe that these sources are reliable, we do not guarantee the accuracy or completeness of this information and we have not independently verified this information. Although we are not aware of any misstatements regarding the market and industry data presented in this prospectus and the documents incorporated herein by reference, these estimates involve risks and uncertainties and are subject to change based on various factors, including those discussed under the heading “Risk Factors” contained in the applicable prospectus supplement and any related free writing prospectus, and under similar headings in the other documents that are incorporated by reference into this prospectus. Accordingly, investors should not place undue reliance on this information.
This prospectus contains summaries of certain provisions contained in some of the documents described herein, but reference is made to the actual documents for complete information. All of the summaries are qualified in their entirety by the actual documents. Copies of some of the documents referred to herein have been filed, will be filed or will be incorporated by reference as exhibits to the registration statement of which this prospectus is a part, and you may obtain copies of those documents as described below under the section titled “Where You Can Find Additional Information.”
Table of Contents
This summary highlights selected information appearing elsewhere in this prospectus or incorporated by reference in this prospectus, and does not contain all of the information that you need to consider in making your investment decision. You should carefully read this entire prospectus, the applicable prospectus supplement and any related free writing prospectus, including the risks of investing in our securities discussed under the heading “Risk Factors” contained in the applicable prospectus supplement and any related free writing prospectus, and under similar headings in the other documents that are incorporated by reference into this prospectus. You should also carefully read the other information incorporated by reference into this prospectus, including our financial statements, and the exhibits to the registration statement of which this prospectus is a part.
NGM Biopharmaceuticals, Inc.
Overview
We are a biopharmaceutical company focused on developing novel therapeutics based on our scientific understanding of key biological pathways underlying cardio-metabolic, liver, oncologic and ophthalmic diseases. These diseases represent leading causes of morbidity and mortality and a significant burden for healthcare systems. Since the commencement of our operations in 2008, we have generated a robust portfolio of product candidates. We have created this portfolio using our research and drug discovery approach that employs unbiased, in vivo-based discovery to identify proprietary insights into critical biological processes. We combine this approach with our protein and antibody engineering expertise to find the appropriate modality to enhance each product candidate’s therapeutic potential.
Our most advanced programs are focused on novel discoveries in hormone pathways that regulate cardio-metabolic processes and liver function, including those driving NASH, type 2 diabetes and obesity. We have identified multiple hormone pathways of interest, the most advanced of which are: FGF19, which plays a critical role in controlling bile acid, lipid and glucose metabolism; FGFR1c/KLB, which regulates insulin sensitivity, blood glucose and liver fat; and GDF15, which drives profound metabolic activity by regulating fuel flux and has been considered a challenging therapeutic target. We believe these hormone pathways work through distinct mechanisms and play an important role in metabolic regulation. Our five most advanced proprietary product candidates are summarized below.
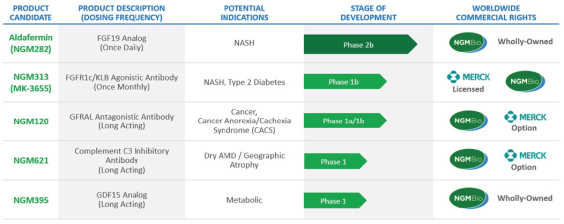
1
Table of Contents
Corporate Information
We were incorporated in Delaware in December 2007 and commenced operations in 2008. Our principal executive offices are located at 333 Oyster Point Blvd., South San Francisco, CA 94080-7014, and our telephone number is (650) 243-5555. Our website address is http://www.ngmbio.com. The contents of and information accessible through our website are not incorporated into this prospectus and our reference to the URL for our website is intended to be an inactive textual reference only.
As used in this prospectus, “NGM Biopharmaceuticals”, “NGM”, “we”, “us” and “our” refer to NGM Biopharmaceuticals, Inc. and its wholly-owned subsidiary taken as a whole. This prospectus and the information incorporated herein by reference include trademarks, service marks and trade names owned by us or other companies. All trademarks, service marks and trade names included or incorporated by reference into this prospectus, any applicable prospectus supplement or any related free writing prospectus are the property of their respective owners. We do not intend our use or display of other companies’ trademarks or trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies or products.
Our common stock is currently listed on The Nasdaq Global Select Market under the symbol “NGM.”
The Securities We May Offer
We may offer shares of our common stock and preferred stock, various series of debt securities and/or warrants to purchase any of such securities, either individually or in combination, from time to time under this prospectus, together with the applicable prospectus supplement and any related free writing prospectus, at prices and on terms to be determined by market conditions at the time of any offering. There is no limit on the aggregate amount of the securities that we may offer pursuant to the registration statement of which this prospectus is a part. We may also offer common stock, preferred stock and/or debt securities upon the exercise of warrants. This prospectus provides you with a general description of the securities we may offer. Each time we offer a type or series of securities under this prospectus, we will provide a prospectus supplement that will describe the specific amounts, prices and other important terms of the securities, including, to the extent applicable:
| ∎ | designation or classification; |
| ∎ | aggregate principal amount or aggregate offering price; |
| ∎ | maturity date, if applicable; |
| ∎ | original issue discount, if any; |
| ∎ | rates and times of payment of interest or dividends, if any; |
| ∎ | redemption, conversion, exercise, exchange or sinking fund terms, if any; |
| ∎ | conversion or exchange prices or rates, if any, and, if applicable, any provisions for changes to or adjustments in the conversion or exchange prices or rates and in the securities or other property receivable upon conversion or exchange; |
| ∎ | ranking; |
| ∎ | restrictive covenants, if any; |
| ∎ | voting or other rights, if any; and |
| ∎ | material or special U.S. federal income tax considerations, if any. |
The applicable prospectus supplement and any related free writing prospectus that we may authorize to be provided to you may also add, update or change any of the information contained in this prospectus or in the documents we have incorporated by reference. However, no prospectus supplement or free writing prospectus will offer a security that is not registered and described in this prospectus at the time of the effectiveness of the registration statement of which this prospectus is a part.
2
Table of Contents
THIS PROSPECTUS MAY NOT BE USED TO CONSUMMATE A SALE OF SECURITIES UNLESS IT IS ACCOMPANIED BY A PROSPECTUS SUPPLEMENT.
We may sell the securities directly to investors or to or through agents, underwriters or dealers. We and our agents or underwriters reserve the right to accept or reject all or part of any proposed purchase of securities. If we do offer securities to or through agents or underwriters, we will include in the applicable prospectus supplement:
| ∎ | the names of those agents or underwriters; |
| ∎ | applicable fees, discounts and commissions to be paid to them; |
| ∎ | details regarding any options under which underwriters may purchase additional securities from us, if any; and |
| ∎ | the net proceeds to us. |
Common Stock. We may issue shares of our common stock from time to time. The holders of our common stock are entitled to one vote for each share held of record on all matters submitted to a vote of stockholders. Subject to preferences that may be applicable to any outstanding preferred stock, the holders of common stock are entitled to receive ratably any dividends that our board of directors may declare out of funds legally available for that purpose. In the event of our liquidation, dissolution or winding up, holders of our common stock are entitled to share ratably in all assets remaining after payment of liabilities and the liquidation preference of any outstanding preferred stock. Holders of our common stock have no preemptive, conversion, subscription or other rights, and there are no redemption or sinking fund provisions applicable to our common stock. In this prospectus, we have summarized certain general features of the common stock under “Description of Capital Stock — Common Stock.” We urge you, however, to read the applicable prospectus supplement (and any related free writing prospectus that we may authorize to be provided to you) related to any common stock being offered.
Preferred Stock. We may issue shares of our preferred stock from time to time, in one or more series. If we sell any series of preferred stock under this prospectus, our board of directors will fix the designations, voting powers, preferences and rights of the preferred stock of each series we issue under this prospectus, as well as the qualifications, limitations or restrictions thereof, including dividend rights, conversion rights, preemptive rights, terms of redemption or repurchase, liquidation preferences, sinking fund terms and the number of shares constituting any series or the designation of any series, in the certificate of designation relating to that series. Preferred stock may be convertible into our common stock or other securities of ours, or may be exchangeable for debt securities. Conversion may be mandatory or at the holder’s option and would be at prescribed conversion rates. We will file as an exhibit to the registration statement of which this prospectus is a part, or will incorporate by reference from reports that we file with the SEC, the form of any certificate of designation that contains the terms of the series of preferred stock we are offering. In this prospectus, we have summarized certain general features of the preferred stock under “Description of Capital Stock — Preferred Stock.” We urge you, however, to read the applicable prospectus supplement (and any related free writing prospectus that we may authorize to be provided to you) related to the series of preferred stock being offered, as well as the complete certificate of designation that contains the terms of the applicable series of preferred stock.
Debt Securities. We may issue debt securities from time to time, in one or more series, as either senior or subordinated debt or as senior or subordinated convertible debt. The senior debt securities will rank equally with any other unsecured and unsubordinated debt. The subordinated debt securities will be subordinate and junior in right of payment, to the extent and in the manner described in the instrument governing the debt, to all of our senior indebtedness. Convertible debt securities will be convertible into or exchangeable for our common stock or other securities. Conversion may be mandatory or at your option and would be at prescribed conversion rates.
3
Table of Contents
Any debt securities issued under this prospectus will be issued under one or more documents called indentures, which are contracts between us and a national banking association or other eligible party, as trustee. In this prospectus, we have summarized certain general features of the debt securities under “Description of Debt Securities.” We urge you, however, to read the applicable prospectus supplement (and any free writing prospectus that we may authorize to be provided to you) related to the series of debt securities being offered, as well as the complete indentures that contain the terms of the debt securities. We have filed the form of indenture as an exhibit to the registration statement of which this prospectus is a part, and supplemental indentures and forms of debt securities containing the terms of the debt securities being offered will be filed as exhibits to the registration statement of which this prospectus is a part or will be incorporated by reference from reports that we file with the SEC.
Warrants. We may issue warrants for the purchase of common stock, preferred stock and/or debt securities in one or more series. We may issue warrants independently or in combination with common stock, preferred stock and/or debt securities. In this prospectus, we have summarized certain general features of the warrants under “Description of Warrants.” We urge you, however, to read the applicable prospectus supplement (and any related free writing prospectus that we may authorize to be provided to you) related to the particular series of warrants being offered, as well as any warrant agreements and warrant certificates that contain the terms of the warrants. We have filed forms of the warrant agreements and forms of warrant certificates containing the terms of the warrants that may be offered as exhibits to the registration statement of which this prospectus is a part. We will file as exhibits to the registration statement of which this prospectus is a part, or will incorporate by reference from reports that we file with the SEC, the form of warrant and/or the warrant agreement and warrant certificate, as applicable, that contain the terms of the particular series of warrants we are offering, and any supplemental agreements, before the issuance of such warrants.
Any warrants issued under this prospectus may be evidenced by warrant certificates. Warrants also may be issued under an applicable warrant agreement that we enter into with a warrant agent. We will indicate the name and address of the warrant agent, if applicable, in the prospectus supplement relating to the particular series of warrants being offered.
Use of Proceeds
Except as described in any applicable prospectus supplement or in any free writing prospectuses we have authorized for use in connection with a specific offering, we currently intend to use the net proceeds from the sale of the securities offered by us hereunder, if any, for working capital and other general corporate purposes, which may include funding research and development, general and administrative activities and capital expenditures. See “Use of Proceeds” in this prospectus.
Nasdaq Global Select Market Listing
Our common stock is listed on The Nasdaq Global Select Market under the symbol “NGM.” The applicable prospectus supplement will contain information, where applicable, as to other listings, if any, on The Nasdaq Global Select Market or other securities exchange of the securities covered by the applicable prospectus supplement.
4
Table of Contents
Investing in our securities involves a high degree of risk. Before deciding whether to invest in our securities, you should consider carefully the risks and uncertainties described under the heading “Risk Factors” contained in the applicable prospectus supplement and any related free writing prospectus, and discussed under the section titled “Risk Factors” contained in our most recent Annual Report on Form 10-K and in our most recent Quarterly Report on Form 10-Q, as well as any amendments thereto reflected in subsequent filings with the SEC, which are incorporated by reference into this prospectus in their entirety, together with other information in this prospectus, any applicable prospectus supplement, the documents incorporated by reference and any free writing prospectus that we may authorize for use in connection with this offering. The risks described in these documents are not the only ones we face, but those that we consider to be material. There may be other unknown or unpredictable economic, business, competitive, regulatory or other factors that could have material adverse effects on our future results. Past financial performance may not be a reliable indicator of future performance, and historical trends should not be used to anticipate results or trends in future periods. If any of these risks actually occurs, our business, financial condition, results of operations or cash flow could be seriously harmed. This could cause the trading price of our common stock to decline, resulting in a loss of all or part of your investment. Please also read carefully the section below titled “Special Note Regarding Forward-Looking Statements.”
5
Table of Contents
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents we have filed with the SEC that are incorporated by reference contain “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, or the Exchange Act. These statements relate to future events or to our future operating or financial performance and involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. Forward-looking statements may include, but are not limited to, statements about:
| ∎ | the success, cost and timing of our product development activities and clinical trials; |
| ∎ | our or our partners’ ability to obtain and maintain regulatory approval for aldafermin (NGM282), NGM313 (MK-3655), NGM120, NGM621, NGM395 and any of our future product candidates, and any related restrictions, limitations, and/or warnings in the label of an approved product candidate; |
| ∎ | our belief that aldafermin may have the potential to be a treatment for non-alcoholic steatohepatitis, or NASH, patients with moderate to advanced fibrosis; |
| ∎ | our belief regarding the impact of our product candidate side effects and our ability to effectively manage these side effects; |
| ∎ | our belief that MK-3655 (formerly NGM313) may have the potential to be a treatment for NASH patients with early to moderate fibrosis with or without type 2 diabetes; |
| ∎ | the potential renewal of our research collaboration, product development and license agreement with Merck Sharp & Dohme Corp., or Merck, and the possibility that Merck will decide to exercise its option to license certain programs upon our completion of a proof-of-concept study in humans; |
| ∎ | our ability to obtain funding for our operations; |
| ∎ | the commercialization of our product candidates, if approved; |
| ∎ | our plans to research, develop and commercialize our product candidates; |
| ∎ | our ability to attract additional collaborators with development, regulatory and commercialization expertise; |
| ∎ | current and future agreements with third parties in connection with the commercialization of aldafermin, NGM313, NGM120, NGM621, NGM395 or any other future approved product; |
| ∎ | the size and growth potential of the markets for our product candidates, and our ability to serve those markets; |
| ∎ | the rate and degree of market acceptance of our product candidates, as well as the reimbursement coverage for our product candidates; |
| ∎ | regulatory developments in the United States and foreign countries; |
| ∎ | the performance of, and our ability to obtain sufficient supply of clinical trial materials in a timely manner from, third-party suppliers and manufacturers; |
| ∎ | the success of competing therapies that are or may become available; |
| ∎ | our ability to attract and retain key scientific, development and management personnel; |
| ∎ | our estimates regarding future expenses, revenue, capital requirements and needs for additional financing; |
| ∎ | our expectations regarding the period during which we qualify as an emerging growth company under the Jumpstart Our Business Startups Act of 2012, as amended; |
| ∎ | our expectations regarding our ability to obtain, maintain, protect and enforce intellectual property protection for our product candidates; and |
| ∎ | our anticipated use of the net proceeds from offerings of our securities under this prospectus. |
In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,” “would,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “projects,” “predicts,” “potential” and similar expressions intended to identify forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and are subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. We discuss in greater detail many of these
6
Table of Contents
risks under the heading “Risk Factors” contained in the applicable prospectus supplement, in any free writing prospectuses we may authorize for use in connection with a specific offering, and in our most recent Annual Report on Form 10-K and in our most recent Quarterly Report on Form 10-Q, as well as any amendments thereto reflected in subsequent filings with the SEC, which are incorporated by reference into this prospectus in their entirety. Also, these forward-looking statements represent our estimates and assumptions only as of the date of the document containing the applicable statement. Unless required by law, we undertake no obligation to update or revise any forward-looking statements to reflect new information or future events or developments. Thus, you should not assume that our silence over time means that actual events are bearing out as expressed or implied in such forward-looking statements. You should read this prospectus, the applicable prospectus supplement, together with the documents we have filed with the SEC that are incorporated by reference and any free writing prospectus that we may authorize for use in connection with a specific offering completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of the forward-looking statements in the foregoing documents by these cautionary statements.
7
Table of Contents
Except as described in any applicable prospectus supplement or in any free writing prospectuses we have authorized for use in connection with a specific offering, we currently intend to use the net proceeds from the sale of the securities offered by us hereunder, if any, for working capital and other general corporate purposes, which may include funding research and development, general and administrative activities and capital expenditures.
8
Table of Contents
General
As of the date of this prospectus, our authorized capital stock consists of 400,000,000 shares of common stock, $0.001 par value per share, and 10,000,000 shares of preferred stock, $0.001 par value per share.
The following summary description of our capital stock is based on the provisions of our amended and restated certificate of incorporation, our amended and restated bylaws and applicable provisions of the General Corporation Law of the State of Delaware. The description is intended as a summary, and is qualified in its entirety by reference to our amended and restated certificate of incorporation, our amended and restated bylaws and the General Corporation Law of the State of Delaware. For information on how to obtain copies of our amended and restated certificate of incorporation and our amended and restated bylaws, which are exhibits to the registration statement of which this prospectus is a part, see “Where You Can Find Additional Information.”
Common Stock
Voting Rights
Each holder of common stock is entitled to one vote for each share on all matters submitted to a vote of the stockholders. In addition to any vote of holders of a specific class or series, or required by law or the amended and restated certificate of incorporation, the affirmative vote of holders of at least 66 2/3% of the voting power of all of the then outstanding shares of capital stock, voting as a single class, is required to amend certain provisions of our amended and restated certificate of incorporation, including provisions relating to amending our amended and restated bylaws, the classified board, the size of our board, removal of directors, director liability, vacancies on our board, special meetings, stockholder notices, actions by written consent and exclusive jurisdiction.
Dividends
Subject to preferences that may apply to any outstanding preferred stock, holders of our common stock are entitled to receive ratably any dividends that our board of directors may declare out of funds legally available for that purpose.
Liquidation
In the event of our liquidation, dissolution or winding up, holders of our common stock are entitled to share ratably in all assets remaining after payment of liabilities and the liquidation preference of any outstanding preferred stock.
Rights and Preferences
Holders of our common stock have no preemptive, conversion, subscription or other rights, and there are no redemption or sinking fund provisions applicable to our common stock. The rights, preferences and privileges of the holders of our common stock are subject to and may be adversely affected by the rights of the holders of shares of any series of our preferred stock that we may designate in the future.
Fully Paid and Nonassessable
When we issue shares of common stock under this prospectus, the shares will be fully paid and nonassessable.
Preferred Stock
Our board of directors has the authority, without further action by our stockholders, to issue up to 10,000,000 shares of preferred stock in one or more series and to fix the number, rights, preferences, privileges and restrictions thereof. These rights, preferences and privileges could include dividend rights, conversion rights, voting rights, terms of redemption, liquidation preferences and sinking fund terms, and the number of shares constituting any series or the designation of such series, any or all of which may be greater than the rights of common stock.
Our board of directors will fix the designations, voting powers, preferences and rights of the preferred stock of each series we issue under this prospectus, as well as the qualifications, limitations or restrictions thereof, in the
9
Table of Contents
certificate of designation relating to that series. We will file as an exhibit to the registration statement of which this prospectus is a part, or will incorporate by reference from reports that we file with the SEC, the form of any certificate of designation that contains the terms of the series of preferred stock we are offering. We will describe in the applicable prospectus supplement the terms of the series of preferred stock being offered, including, to the extent applicable:
| ∎ | the title and stated value; |
| ∎ | the number of shares we are offering; |
| ∎ | the liquidation preference per share; |
| ∎ | the purchase price; |
| ∎ | the dividend rate, period and payment date and method of calculation for dividends; |
| ∎ | whether dividends will be cumulative or non-cumulative and, if cumulative, the date from which dividends will accumulate; |
| ∎ | the procedures for any auction and remarketing; |
| ∎ | the provisions for any sinking fund; |
| ∎ | the provisions for redemption or repurchase, if applicable, and any restrictions on our ability to exercise those redemption and repurchase rights; |
| ∎ | any listing of the preferred stock on any securities exchange or market; |
| ∎ | whether the preferred stock will be convertible into our common stock, and, if applicable, the conversion price, or how it will be calculated, and the conversion period; |
| ∎ | whether the preferred stock will be exchangeable into debt securities, and, if applicable, the exchange price, or how it will be calculated, and the exchange period; |
| ∎ | voting rights of the preferred stock; |
| ∎ | preemptive rights; |
| ∎ | restrictions on transfer, sale or other assignment; |
| ∎ | whether interests in the preferred stock will be represented by depositary shares; |
| ∎ | a discussion of material U.S. federal income tax considerations applicable to the preferred stock; |
| ∎ | the relative ranking and preferences of the preferred stock as to dividend rights and rights if we liquidate, dissolve or wind up our affairs; |
| ∎ | any limitations on the issuance of any class or series of preferred stock ranking senior to or on a parity with the series of preferred stock as to dividend rights and rights if we liquidate, dissolve or wind up our affairs; and |
| ∎ | any other specific terms, preferences, rights or limitations of, or restrictions on, the preferred stock. |
When we issue shares of preferred stock under this prospectus, the shares will be fully paid and non-assessable.
Unless we specify otherwise in the applicable prospectus supplement, the preferred stock will rank, with respect to dividends and upon our liquidation, dissolution or winding up:
| ∎ | senior to all classes or series of our common stock and to all of our equity securities ranking junior to the preferred stock; |
| ∎ | on a parity with all of our equity securities the terms of which specifically provide that the equity securities rank on a parity with the preferred stock; and |
| ∎ | junior to all of our equity securities the terms of which specifically provide that the equity securities rank senior to the preferred stock. |
The term “equity securities” does not include convertible debt securities.
The General Corporation Law of the State of Delaware, the state of our incorporation, provides that the holders of preferred stock will have the right to vote separately as a class on any proposal involving fundamental changes in the rights of holders of that preferred stock. This right is in addition to any voting rights that may be provided for in the applicable certificate of designation.
10
Table of Contents
The issuance of our preferred stock could adversely affect the voting power of holders of common stock and the likelihood that such holders will receive dividend payments and payments upon liquidation. In addition, the issuance of preferred stock could have the effect of delaying, deferring or preventing a change in control or other corporate action.
Anti-takeover Effects of Provisions of our Certificate of Incorporation and Bylaws and Delaware Law
Certificate of Incorporation and Bylaws
Our amended and restated certificate of incorporation and amended and restated bylaws include a number of provisions that may deter or impede hostile takeovers or changes of control or management. These provisions include:
| ∎ | Issuance of Undesignated Preferred Stock: Our board of directors has the authority, without further action by the stockholders, to issue up to 10,000,000 shares of undesignated preferred stock with rights and preferences, including voting rights, designated from time to time by our board of directors. The existence of authorized but unissued shares of preferred stock enables our board of directors to make it more difficult to attempt to obtain control of us by means of a merger, tender offer, proxy contest or otherwise. |
| ∎ | Classified Board: Our amended and restated certificate of incorporation provides for a classified board of directors consisting of three classes of directors, with staggered three-year terms. Only one class of directors will be elected at each annual meeting of our stockholders, with the other classes continuing for the remainder of their respective three-year terms. This provision may have the effect of delaying a change in control of our board. |
| ∎ | Board of Directors Vacancies: Our amended and restated certificate of incorporation and amended and restated bylaws authorize only our board of directors to fill vacant directorships, subject to the rights of the holders of any series of preferred stock or as otherwise provided by applicable law. In addition, the number of directors constituting our board of directors may be set only by resolution adopted by a majority vote of our entire board of directors. These provisions prevent a stockholder from increasing the size of our board of directors and gaining control of our board of directors by filling the resulting vacancies with its own nominees. |
| ∎ | Stockholder Action; Special Meetings of Stockholders: Our amended and restated certificate of incorporation provides that our stockholders may not take action by written consent, but may only take action at annual or special meetings of our stockholders. Stockholders are not permitted to cumulate their votes for the election of directors. Our amended and restated bylaws provide that only the chairman of our board of directors, our chief executive officer, or a majority of our board of directors may call special meetings of our stockholders. |
| ∎ | Advance Notice Requirements for Stockholder Proposals and Director Nominations: Our amended and restated bylaws provide advance notice procedures for stockholders seeking to bring business before our annual meeting of stockholders, or to nominate candidates for election as directors at our annual meeting of stockholders. Our amended and restated bylaws also specify certain requirements as to the form and content of a stockholder’s notice. These provisions may make it more difficult for our stockholders to bring matters before our annual meeting of stockholders or to nominate directors at annual meetings of stockholders. |
We designed these provisions to enhance the likelihood of continued stability in the composition of our board of directors and its policies, to discourage certain types of transactions that may involve an actual or threatened acquisition of us and to reduce our vulnerability to an unsolicited acquisition proposal. We also designed these provisions to discourage certain tactics that may be used in proxy fights. However, these provisions could have the effect of discouraging others from making tender offers for our shares and, as a consequence, they may also reduce fluctuations in the market price of our shares that could result from actual or rumored takeover attempts.
11
Table of Contents
Section 203 of the Delaware General Corporation Law
We are subject to Section 203 of the General Corporation Law of the State of Delaware, or the DGCL, which prohibits a Delaware corporation from engaging in a business combination with any interested stockholder for a period of three years following the date the person became an interested stockholder, with the following exceptions:
| ∎ | before such date, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested holder; |
| ∎ | upon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction began, excluding for purposes of determining the voting stock outstanding (but not the outstanding voting stock owned by the interested stockholder) those shares owned (a) by persons who are directors and also officers and (b) pursuant to employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; and |
| ∎ | on or after such date, the business combination is approved by the board of directors and authorized at an annual or special meeting of the stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock that is not owned by the interested stockholder. |
In general, Section 203 defines business combination to include the following:
| ∎ | any merger or consolidation involving the corporation and the interested stockholder; |
| ∎ | any sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation involving the interested stockholder; |
| ∎ | subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; |
| ∎ | any transaction involving the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; or |
| ∎ | the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits by or through the corporation. |
In general, Section 203 of the DGCL defines an interested stockholder as an entity or person who, together with the entity’s or person’s affiliates and associates, beneficially owns, or is an affiliate of the corporation and within three years prior to the time of determination of interested stockholder status did own, 15% or more of the outstanding voting stock of the corporation.
A Delaware corporation may opt out of these provisions with an express provision in its certificate of incorporation. We have not opted out of these provisions, which may, as a result, discourage or prevent mergers or other takeover or change in control attempts of us.
Choice of Forum
Our amended and restated certificate of incorporation provides that the Court of Chancery of the State of Delaware will be the exclusive forum for the following types of actions or proceedings under Delaware statutory or common law: (i) any derivative action or proceeding brought on our behalf; (ii) any action asserting a breach of fiduciary duty owed by any director, officer or other employee to us or our stockholders; (iii) any action asserting a claim against us or any director or officer or other employee arising pursuant to the Delaware General Corporation Law, our amended and restated certificate of incorporation or amended and restated bylaws; or (iv) any action asserting a claim against us or any director or officer or other employee that is governed by the internal affairs doctrine. This provision would not apply to claims brought to enforce a duty or liability created by the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. Our amended and restated certificate of incorporation further provides that the federal district courts of the United States of America will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act.
12
Table of Contents
Transfer Agent and Registrar
Our transfer agent and registrar for our common stock is American Stock Transfer & Trust Company, LLC.
Listing on the Nasdaq Global Select Market
Our common stock is listed on The Nasdaq Global Select Market under the symbol “NGM.”
13
Table of Contents
DESCRIPTION OF DEBT SECURITIES
We may issue debt securities from time to time, in one or more series, as either senior or subordinated debt or as senior or subordinated convertible debt. While the terms we have summarized below will apply generally to any debt securities that we may offer under this prospectus, we will describe the particular terms of any debt securities that we may offer in more detail in the applicable prospectus supplement. The terms of any debt securities offered under a prospectus supplement may differ from the terms described below. Unless the context requires otherwise, whenever we refer to the indenture, we also are referring to any supplemental indentures that specify the terms of a particular series of debt securities.
We will issue the debt securities under the indenture that we will enter into with the trustee named in the indenture. The indenture will be qualified under the Trust Indenture Act of 1939, as amended, or the Trust Indenture Act. We have filed the form of indenture as an exhibit to the registration statement of which this prospectus is a part, and supplemental indentures and forms of debt securities containing the terms of the debt securities being offered will be filed as exhibits to the registration statement of which this prospectus is a part or will be incorporated by reference from reports that we file with the SEC.
The following summary of material provisions of the debt securities and the indenture is subject to, and qualified in its entirety by reference to, all of the provisions of the indenture applicable to a particular series of debt securities. We urge you to read the applicable prospectus supplements and any related free writing prospectuses related to the debt securities that we may offer under this prospectus, as well as the complete indenture that contains the terms of the debt securities.
General
The indenture does not limit the amount of debt securities that we may issue. It provides that we may issue debt securities up to the principal amount that we may authorize and may be in any currency or currency unit that we may designate. Except for the limitations on consolidation, merger and sale of all or substantially all of our assets contained in the indenture, the terms of the indenture do not contain any covenants or other provisions designed to give holders of any debt securities protection against changes in our operations, financial condition or transactions involving us.
We may issue the debt securities issued under the indenture as “discount securities,” which means they may be sold at a discount below their stated principal amount. These debt securities, as well as other debt securities that are not issued at a discount, may be issued with “original issue discount,” or OID, for U.S. federal income tax purposes because of interest payment and other characteristics or terms of the debt securities. Material U.S. federal income tax considerations applicable to debt securities issued with OID will be described in more detail in any applicable prospectus supplement.
We will describe in the applicable prospectus supplement the terms of the series of debt securities being offered, including:
| ∎ | the title of the series of debt securities; |
| ∎ | any limit upon the aggregate principal amount that may be issued; |
| ∎ | the maturity date or dates; |
| ∎ | the form of the debt securities of the series; |
| ∎ | the applicability of any guarantees; |
| ∎ | whether or not the debt securities will be secured or unsecured, and the terms of any secured debt; |
| ∎ | whether the debt securities rank as senior debt, senior subordinated debt, subordinated debt or any combination thereof, and the terms of any subordination; |
| ∎ | if the price (expressed as a percentage of the aggregate principal amount thereof) at which such debt securities will be issued is a price other than the principal amount thereof, the portion of the principal amount thereof payable upon declaration of acceleration of the maturity thereof, or if applicable, the portion |
14
Table of Contents
| of the principal amount of such debt securities that is convertible into another security or the method by which any such portion shall be determined; |
| ∎ | the interest rate or rates, which may be fixed or variable, or the method for determining the rate and the date interest will begin to accrue, the dates interest will be payable and the regular record dates for interest payment dates or the method for determining such dates; |
| ∎ | our right, if any, to defer payment of interest and the maximum length of any such deferral period; |
| ∎ | if applicable, the date or dates after which, or the period or periods during which, and the price or prices at which, we may, at our option, redeem the series of debt securities pursuant to any optional or provisional redemption provisions and the terms of those redemption provisions; |
| ∎ | the date or dates, if any, on which, and the price or prices at which, we are obligated, pursuant to any mandatory sinking fund or analogous fund provisions or otherwise, to redeem, or at the holder’s option to purchase, the series of debt securities and the currency or currency unit in which the debt securities are payable; |
| ∎ | the denominations in which we will issue the series of debt securities, if other than denominations of $1,000 and any integral multiple thereof; |
| ∎ | any and all terms, if applicable, relating to any auction or remarketing of the debt securities of that series and any security for our obligations with respect to such debt securities and any other terms which may be advisable in connection with the marketing of debt securities of that series; |
| ∎ | whether the debt securities of the series shall be issued in whole or in part in the form of a global security or securities; |
| ∎ | the terms and conditions, if any, upon which such global security or securities may be exchanged in whole or in part for other individual securities, and the depositary for such global security or securities; |
| ∎ | if applicable, the provisions relating to conversion or exchange of any debt securities of the series and the terms and conditions upon which such debt securities will be so convertible or exchangeable, including the conversion or exchange price, as applicable, or how it will be calculated and may be adjusted, any mandatory or optional (at our option or the holders’ option) conversion or exchange features, the applicable conversion or exchange period and the manner of settlement for any conversion or exchange; |
| ∎ | if other than the full principal amount thereof, the portion of the principal amount of debt securities of the series which shall be payable upon declaration of acceleration of the maturity thereof; |
| ∎ | additions to or changes in the covenants applicable to the particular debt securities being issued, including, among others, the consolidation, merger or sale covenant; |
| ∎ | additions to or changes in the events of default with respect to the securities and any change in the right of the trustee or the holders to declare the principal, premium, if any, and interest, if any, with respect to such securities to be due and payable; |
| ∎ | additions to or changes in or deletions of the provisions relating to covenant defeasance and legal defeasance; |
| ∎ | additions to or changes in the provisions relating to satisfaction and discharge of the indenture; |
| ∎ | additions to or changes in the provisions relating to the modification of the indenture both with and without the consent of holders of debt securities issued under the indenture; |
| ∎ | the currency of payment of debt securities if other than U.S. dollars and the manner of determining the equivalent amount in U.S. dollars; |
| ∎ | whether interest will be payable in cash or additional debt securities at our or the holders’ option and the terms and conditions upon which the election may be made; |
| ∎ | the terms and conditions, if any, upon which we will pay amounts in addition to the stated interest, premium, if any, and principal amounts of the debt securities of the series to any holder that is not a “United States person” for federal tax purposes; |
| ∎ | any restrictions on transfer, sale or assignment of the debt securities of the series; and |
| ∎ | any other specific terms, preferences, rights or limitations of, or restrictions on, the debt securities, any other additions or changes in the provisions of the indenture, and any terms that may be required by us or advisable under applicable laws or regulations. |
15
Table of Contents
Conversion or Exchange Rights
We will set forth in the applicable prospectus supplement the terms on which a series of debt securities may be convertible into or exchangeable for our common stock or our other securities. We will include provisions as to settlement upon conversion or exchange and whether conversion or exchange is mandatory, at the option of the holder or at our option. We may include provisions pursuant to which the number of shares of our common stock or our other securities that the holders of the series of debt securities receive would be subject to adjustment.
Consolidation, Merger or Sale
Unless we provide otherwise in the prospectus supplement applicable to a particular series of debt securities, the indenture will not contain any covenant that restricts our ability to merge or consolidate, or sell, convey, transfer or otherwise dispose of our assets as an entirety or substantially as an entirety. However, any successor to or acquirer of such assets (other than a subsidiary of ours) must assume all of our obligations under the indenture or the debt securities, as appropriate.
Events of Default under the Indenture
Unless we provide otherwise in the prospectus supplement applicable to a particular series of debt securities, the following are events of default under the indenture with respect to any series of debt securities that we may issue:
| ∎ | if we fail to pay any installment of interest on any series of debt securities, as and when the same shall become due and payable, and such default continues for a period of 90 days; provided, however, that a valid extension of an interest payment period by us in accordance with the terms of any indenture supplemental thereto shall not constitute a default in the payment of interest for this purpose; |
| ∎ | if we fail to pay the principal of, or premium, if any, on any series of debt securities as and when the same shall become due and payable whether at maturity, upon redemption, by declaration or otherwise, or in any payment required by any sinking or analogous fund established with respect to such series; provided, however, that a valid extension of the maturity of such debt securities in accordance with the terms of any indenture supplemental thereto shall not constitute a default in the payment of principal or premium, if any; |
| ∎ | if we fail to observe or perform any other covenant or agreement contained in the debt securities or the indenture, other than a covenant specifically relating to another series of debt securities, and our failure continues for 90 days after we receive written notice of such failure, requiring the same to be remedied and stating that such is a notice of default thereunder, from the trustee or holders of at least 25% in aggregate principal amount of the outstanding debt securities of the applicable series; and |
| ∎ | if specified events of bankruptcy, insolvency or reorganization occur. |
If an event of default with respect to debt securities of any series occurs and is continuing, other than an event of default specified in the last bullet point above, the trustee or the holders of at least 25% in aggregate principal amount of the outstanding debt securities of that series, by notice to us in writing, and to the trustee if notice is given by such holders, may declare the unpaid principal of, premium, if any, and accrued interest, if any, due and payable immediately. If an event of default specified in the last bullet point above occurs with respect to us, the principal amount of and accrued interest, if any, of each issue of debt securities then outstanding shall be due and payable without any notice or other action on the part of the trustee or any holder.
The holders of a majority in principal amount of the outstanding debt securities of an affected series may waive any default or event of default with respect to the series and its consequences, except defaults or events of default regarding payment of principal, premium, if any, or interest, unless we have cured the default or event of default in accordance with the indenture. Any waiver shall cure the default or event of default.
Subject to the terms of the indenture, if an event of default under an indenture shall occur and be continuing, the trustee will be under no obligation to exercise any of its rights or powers under such indenture at the request or direction of any of the holders of the applicable series of debt securities, unless such holders have offered the trustee reasonable indemnity. The holders of a majority in principal amount of the outstanding debt securities of any series will have the right to direct the time, method and place of conducting any proceeding for any remedy available
16
Table of Contents
to the trustee, or exercising any trust or power conferred on the trustee, with respect to the debt securities of that series, provided that:
| ∎ | the direction so given by the holder is not in conflict with any law or the applicable indenture; and |
| ∎ | subject to its duties under the Trust Indenture Act, the trustee need not take any action that might involve it in personal liability or might be unduly prejudicial to the holders not involved in the proceeding. |
A holder of the debt securities of any series will have the right to institute a proceeding under the indenture or to appoint a receiver or trustee, or to seek other remedies only if:
| ∎ | the holder has given written notice to the trustee of a continuing event of default with respect to that series; |
| ∎ | the holders of at least 25% in aggregate principal amount of the outstanding debt securities of that series have made written request, |
| ∎ | such holders have offered to the trustee indemnity satisfactory to it against the costs, expenses and liabilities to be incurred by the trustee in compliance with the request; and |
| ∎ | the trustee does not institute the proceeding, and does not receive from the holders of a majority in aggregate principal amount of the outstanding debt securities of that series other conflicting directions within 90 days after the notice, request and offer. |
These limitations do not apply to a suit instituted by a holder of debt securities if we default in the payment of the principal, premium, if any, or interest on the debt securities.
We will periodically file statements with the trustee regarding our compliance with specified covenants in the indenture.
Modification of Indenture; Waiver
We and the trustee may change an indenture without the consent of any holders with respect to specific matters:
| ∎ | to cure any ambiguity, defect or inconsistency in the indenture or in the debt securities of any series; |
| ∎ | to comply with the provisions described above under “Description of Debt Securities-Consolidation, Merger or Sale;” |
| ∎ | to provide for uncertificated debt securities in addition to or in place of certificated debt securities; |
| ∎ | to add to our covenants, restrictions, conditions or provisions such new covenants, restrictions, conditions or provisions for the benefit of the holders of all or any series of debt securities, to make the occurrence, or the occurrence and the continuance, of a default in any such additional covenants, restrictions, conditions or provisions an event of default or to surrender any right or power conferred upon us in the indenture; |
| ∎ | to add to, delete from or revise the conditions, limitations, and restrictions on the authorized amount, terms, or purposes of issue, authentication and delivery of debt securities, as set forth in the indenture; |
| ∎ | to make any change that does not adversely affect the interests of any holder of debt securities of any series in any material respect; |
| ∎ | to provide for the issuance of and establish the form and terms and conditions of the debt securities of any series as provided above under “Description of Debt Securities-General” to establish the form of any certifications required to be furnished pursuant to the terms of the indenture or any series of debt securities, or to add to the rights of the holders of any series of debt securities; |
| ∎ | to evidence and provide for the acceptance of appointment under any indenture by a successor trustee; or |
| ∎ | to comply with any requirements of the SEC in connection with the qualification of any indenture under the Trust Indenture Act. |
17
Table of Contents
In addition, under the indenture, the rights of holders of a series of debt securities may be changed by us and the trustee with the written consent of the holders of at least a majority in aggregate principal amount of the outstanding debt securities of each series that is affected. However, unless we provide otherwise in the prospectus supplement applicable to a particular series of debt securities, we and the trustee may make the following changes only with the consent of each holder of any outstanding debt securities affected:
| ∎ | extending the fixed maturity of any debt securities of any series; |
| ∎ | reducing the principal amount, reducing the rate of or extending the time of payment of interest, or reducing any premium payable upon the redemption of any series of any debt securities; or |
| ∎ | reducing the percentage of debt securities, the holders of which are required to consent to any amendment, supplement, modification or waiver. |
Discharge
Each indenture provides that we can elect to be discharged from our obligations with respect to one or more series of debt securities, except for specified obligations, including obligations to:
| ∎ | provide for payment; |
| ∎ | register the transfer or exchange of debt securities of the series; |
| ∎ | replace stolen, lost or mutilated debt securities of the series; |
| ∎ | pay principal of and premium and interest on any debt securities of the series; |
| ∎ | maintain paying agencies; |
| ∎ | hold monies for payment in trust; |
| ∎ | recover excess money held by the trustee; |
| ∎ | compensate and indemnify the trustee; and |
| ∎ | appoint any successor trustee. |
In order to exercise our rights to be discharged, we must deposit with the trustee money or government obligations sufficient to pay all the principal of, any premium, if any, and interest on, the debt securities of the series on the dates payments are due.
Form, Exchange and Transfer
We will issue the debt securities of each series only in fully registered form without coupons and, unless we provide otherwise in the applicable prospectus supplement, in denominations of $1,000 and any integral multiple thereof. The indenture provides that we may issue debt securities of a series in temporary or permanent global form and as book-entry securities that will be deposited with, or on behalf of, The Depository Trust Company, or DTC, or another depositary named by us and identified in the applicable prospectus supplement with respect to that series. To the extent the debt securities of a series are issued in global form and as book-entry, a description of terms relating to any book-entry securities will be set forth in the applicable prospectus supplement.
At the option of the holder, subject to the terms of the indenture and the limitations applicable to global securities described in the applicable prospectus supplement, the holder of the debt securities of any series can exchange the debt securities for other debt securities of the same series, in any authorized denomination and of like tenor and aggregate principal amount.
Subject to the terms of the indenture and the limitations applicable to global securities set forth in the applicable prospectus supplement, holders of the debt securities may present the debt securities for exchange or for registration of transfer, duly endorsed or with the form of transfer endorsed thereon duly executed if so required by us or the security registrar, at the office of the security registrar or at the office of any transfer agent designated by us for this purpose. Unless otherwise provided in the debt securities that the holder presents for transfer or exchange, we will impose no service charge for any registration of transfer or exchange, but we may require payment of any taxes or other governmental charges.
18
Table of Contents
We will name in the applicable prospectus supplement the security registrar, and any transfer agent in addition to the security registrar, that we initially designate for any debt securities. We may at any time designate additional transfer agents or rescind the designation of any transfer agent or approve a change in the office through which any transfer agent acts, except that we will be required to maintain a transfer agent in each place of payment for the debt securities of each series.
If we elect to redeem the debt securities of any series, we will not be required to:
| ∎ | issue, register the transfer of, or exchange any debt securities of that series during a period beginning at the opening of business 15 days before the day of mailing of a notice of redemption of any debt securities that may be selected for redemption and ending at the close of business on the day of the mailing; or |
| ∎ | register the transfer of or exchange any debt securities so selected for redemption, in whole or in part, except the unredeemed portion of any debt securities we are redeeming in part. |
Information Concerning the Trustee
The trustee, other than during the occurrence and continuance of an event of default under an indenture, undertakes to perform only those duties as are specifically set forth in the applicable indenture. Upon an event of default under an indenture, the trustee must use the same degree of care as a prudent person would exercise or use in the conduct of his or her own affairs. Subject to this provision, the trustee is under no obligation to exercise any of the powers given it by the indenture at the request of any holder of debt securities unless it is offered reasonable security and indemnity against the costs, expenses and liabilities that it might incur.
Payment and Paying Agents
Unless we otherwise indicate in the applicable prospectus supplement, we will make payment of the interest on any debt securities on any interest payment date to the person in whose name the debt securities, or one or more predecessor securities, are registered at the close of business on the regular record date for the interest.
We will pay principal of and any premium and interest on the debt securities of a particular series at the office of the paying agents designated by us, except that unless we otherwise indicate in the applicable prospectus supplement, we will make interest payments by check that we will mail to the holder or by wire transfer to certain holders. Unless we otherwise indicate in the applicable prospectus supplement, we will designate the corporate trust office of the trustee as our sole paying agent for payments with respect to debt securities of each series. We will name in the applicable prospectus supplement any other paying agents that we initially designate for the debt securities of a particular series. We will maintain a paying agent in each place of payment for the debt securities of a particular series.
All money we pay to a paying agent or the trustee for the payment of the principal of or any premium or interest on any debt securities that remains unclaimed at the end of two years after such principal, premium or interest has become due and payable will be repaid to us, and the holder of the debt security thereafter may look only to us for payment thereof.
Governing Law
The indenture and the debt securities will be governed by and construed in accordance with the internal laws of the State of New York, except to the extent that the Trust Indenture Act of 1939 is applicable.
19
Table of Contents
The following description, together with the additional information we may include in any applicable prospectus supplements and in any related free writing prospectuses that we may authorize to be distributed to you, summarizes the material terms and provisions of the warrants that we may offer under this prospectus, which may consist of warrants to purchase common stock, preferred stock or debt securities and be issued in one or more series. Warrants may be offered independently or in combination with common stock, preferred stock or debt securities offered by any prospectus supplement. While the terms we have summarized below will apply generally to any warrants that we may offer under this prospectus, we will describe the particular terms of any series of warrants in more detail in the applicable prospectus supplement. The following description of warrants will apply to the warrants offered by this prospectus unless we provide otherwise in the applicable prospectus supplement. The applicable prospectus supplement for a particular series of warrants may specify different or additional terms.
We have filed forms of the warrant agreements and forms of warrant certificates containing the terms of the warrants that may be offered as exhibits to the registration statement of which this prospectus is a part. We will file as exhibits to the registration statement of which this prospectus is a part, or will incorporate by reference from reports that we file with the SEC, the form of warrant and/or the warrant agreement and warrant certificate, as applicable, that describe the terms of the particular series of warrants we are offering, and any supplemental agreements, before the issuance of such warrants. The following summaries of material terms and provisions of the warrants are subject to, and qualified in their entirety by reference to, all the provisions of the form of warrant and/or the warrant agreement and warrant certificate, as applicable, and any supplemental agreements applicable to a particular series of warrants that we may offer under this prospectus. We urge you to read the applicable prospectus supplement related to the particular series of warrants that we may offer under this prospectus, as well as any related free writing prospectuses, and the complete form of warrant and/or the warrant agreement and warrant certificate, as applicable, and any supplemental agreements, that contain the terms of the warrants.
General
We will describe in the applicable prospectus supplement the terms of the series of warrants being offered, including:
| ∎ | the offering price and aggregate number of warrants offered; |
| ∎ | the currency for which the warrants may be purchased; |
| ∎ | if applicable, the designation and terms of the securities with which the warrants are issued and the number of warrants issued with each such security or each principal amount of such security; |
| ∎ | in the case of warrants to purchase debt securities, the principal amount of debt securities purchasable upon exercise of one warrant and the price at, and currency in which, this principal amount of debt securities may be purchased upon such exercise; |
| ∎ | in the case of warrants to purchase common stock or preferred stock, the number of shares of common stock or preferred stock, as the case may be, purchasable upon the exercise of one warrant and the price at which these shares may be purchased upon such exercise; |
| ∎ | the effect of any merger, consolidation, sale or other disposition of our business on the warrant agreements and the warrants; |
| ∎ | the terms of any rights to redeem or call the warrants; |
| ∎ | any provisions for changes to or adjustments in the exercise price or number of securities issuable upon exercise of the warrants; |
| ∎ | the dates on which the right to exercise the warrants will commence and expire; |
| ∎ | the manner in which the warrant agreements and warrants may be modified; |
| ∎ | a discussion of any material or special U.S. federal income tax considerations of holding or exercising the warrants; |
| ∎ | the terms of the securities issuable upon exercise of the warrants; and |
| ∎ | any other specific terms, preferences, rights or limitations of or restrictions on the warrants. |
20
Table of Contents
| ∎ | Before exercising their warrants, holders of warrants will not have any of the rights of holders of the securities purchasable upon such exercise, including: |
| ∎ | in the case of warrants to purchase debt securities, the right to receive payments of principal of, or premium, if any, or interest on, the debt securities purchasable upon exercise or to enforce covenants in the applicable indenture; or |
| ∎ | in the case of warrants to purchase common stock or preferred stock, the right to receive dividends, if any, or, payments upon our liquidation, dissolution or winding up or to exercise voting rights, if any. |
Exercise of Warrants
Each warrant will entitle the holder to purchase the securities that we specify in the applicable prospectus supplement at the exercise price that we describe in the applicable prospectus supplement. The warrants may be exercised as set forth in the prospectus supplement relating to the warrants offered. Unless we otherwise specify in the applicable prospectus supplement, warrants may be exercised at any time up to the close of business on the expiration date set forth in the prospectus supplement relating to the warrants offered thereby. After the close of business on the expiration date, unexercised warrants will become void.
Upon receipt of payment and the warrant or warrant certificate, as applicable, properly completed and duly executed at the corporate trust office of the warrant agent, if any, or any other office, including ours, indicated in the prospectus supplement, we will, as soon as practicable, issue and deliver the securities purchasable upon such exercise. If less than all of the warrants (or the warrants represented by such warrant certificate) are exercised, a new warrant or a new warrant certificate, as applicable, will be issued for the remaining warrants.
Governing Law
Unless we otherwise specify in the applicable prospectus supplement, the warrants and any warrant agreements will be governed by and construed in accordance with the laws of the State of New York.
Enforceability of Rights by Holders of Warrants
Each warrant agent, if any, will act solely as our agent under the applicable warrant agreement and will not assume any obligation or relationship of agency or trust with any holder of any warrant. A single bank or trust company may act as warrant agent for more than one issue of warrants. A warrant agent will have no duty or responsibility in case of any default by us under the applicable warrant agreement or warrant, including any duty or responsibility to initiate any proceedings at law or otherwise, or to make any demand upon us. Any holder of a warrant may, without the consent of the related warrant agent or the holder of any other warrant, enforce by appropriate legal action its right to exercise, and receive the securities purchasable upon exercise of, its warrants.
21
Table of Contents
We can issue securities in registered form or in the form of one or more global securities. We describe global securities in greater detail below. We refer to those persons who have securities registered in their own names on the books that we or any applicable trustee, depositary or warrant agent maintain for this purpose as the “holders” of those securities. These persons are the legal holders of the securities. We refer to those persons who, indirectly through others, own beneficial interests in securities that are not registered in their own names as “indirect holders” of those securities. As we discuss below, indirect holders are not legal holders, and investors in securities issued in book-entry form or in street name will be indirect holders.
Book-Entry Holders
We may issue securities in book-entry form only, as we will specify in the applicable prospectus supplement. This means securities may be represented by one or more global securities registered in the name of a financial institution that holds them as depositary on behalf of other financial institutions that participate in the depositary’s book-entry system. These participating institutions, which are referred to as participants, in turn, hold beneficial interests in the securities on behalf of themselves or their customers.
Only the person in whose name a security is registered is recognized as the holder of that security. Securities issued in global form will be registered in the name of the depositary or its participants. Consequently, for securities issued in global form, we will recognize only the depositary as the holder of the securities, and we will make all payments on the securities to the depositary. The depositary passes along the payments it receives to its participants, which in turn pass the payments along to their customers who are the beneficial owners. The depositary and its participants do so under agreements they have made with one another or with their customers; they are not obligated to do so under the terms of the securities.
As a result, investors in a book-entry security will not own securities directly. Instead, they will own beneficial interests in a global security, through a bank, broker or other financial institution that participates in the depositary’s book-entry system or holds an interest through a participant. As long as the securities are issued in global form, investors will be indirect holders, and not holders, of the securities.
Street Name Holders
We may terminate a global security or issue securities in non-global form. In these cases, investors may choose to hold their securities in their own names or in “street name.” Securities held by an investor in street name would be registered in the name of a bank, broker or other financial institution that the investor chooses, and the investor would hold only a beneficial interest in those securities through an account he or she maintains at that institution.
For securities held in street name, we will recognize only the intermediary banks, brokers and other financial institutions in whose names the securities are registered as the holders of those securities, and we will make all payments on those securities to them. These institutions pass along the payments they receive to their customers who are the beneficial owners, but only because they agree to do so in their customer agreements or because they are legally required to do so. Investors who hold securities in street name will be indirect holders, not holders, of those securities.
Legal Holders
Our obligations, as well as the obligations of any applicable trustee and of any third parties employed by us or a trustee, run only to the legal holders of the securities. We do not have obligations to investors who hold beneficial interests in global securities, in street name or by any other indirect means. This will be the case whether an investor chooses to be an indirect holder of a security or has no choice because we are issuing the securities only in global form.
For example, once we make a payment or give a notice to the holder, we have no further responsibility for the payment or notice even if that holder is required, under agreements with depositary participants or customers or by law, to pass it along to the indirect holders but does not do so. Similarly, we may want to obtain the approval of the
22
Table of Contents
holders to amend an indenture, to relieve us of the consequences of a default or of our obligation to comply with a particular provision of the indenture or for other purposes. In such an event, we would seek approval only from the holders, and not the indirect holders, of the securities. Whether and how the holders contact the indirect holders is up to the holders.
Special Considerations For Indirect Holders
If you hold securities through a bank, broker or other financial institution, either in book-entry form or in street name, you should check with your own institution to find out:
| ∎ | how it handles securities payments and notices; |
| ∎ | whether it imposes fees or charges; |
| ∎ | how it would handle a request for the holders’ consent, if ever required; |
| ∎ | whether and how you can instruct it to send you securities registered in your own name so you can be a holder, if that is permitted in the future; |
| ∎ | how it would exercise rights under the securities if there were a default or other event triggering the need for holders to act to protect their interests; and |
| ∎ | if the securities are in book-entry form, how the depositary’s rules and procedures will affect these matters. |
Global Securities
A global security is a security that represents one or any other number of individual securities held by a depositary. Generally, all securities represented by the same global securities will have the same terms.
Each security issued in book-entry form will be represented by a global security that we deposit with and register in the name of a financial institution or its nominee that we select. The financial institution that we select for this purpose is called the depositary. Unless we specify otherwise in the applicable prospectus supplement, DTC will be the depositary for all securities issued in book-entry form.
A global security may not be transferred to or registered in the name of anyone other than the depositary, its nominee or a successor depositary, unless special termination situations arise. We describe those situations below under the section titled “Special Situations When a Global Security Will Be Terminated” in this prospectus. As a result of these arrangements, the depositary, or its nominee, will be the sole registered owner and holder of all securities represented by a global security, and investors will be permitted to own only beneficial interests in a global security. Beneficial interests must be held by means of an account with a broker, bank or other financial institution that in turn has an account with the depositary or with another institution that does. Thus, an investor whose security is represented by a global security will not be a holder of the security, but only an indirect holder of a beneficial interest in the global security.
If the prospectus supplement for a particular security indicates that the security will be issued in global form only, then the security will be represented by a global security at all times unless and until the global security is terminated. If termination occurs, we may issue the securities through another book-entry clearing system or decide that the securities may no longer be held through any book-entry clearing system.
Special Considerations For Global Securities
The rights of an indirect holder relating to a global security will be governed by the account rules of the investor’s financial institution and of the depositary, as well as general laws relating to securities transfers. We do not recognize an indirect holder as a holder of securities and instead deal only with the depositary that holds the global security.
If securities are issued only in the form of a global security, an investor should be aware of the following:
| ∎ | an investor cannot cause the securities to be registered in his or her name, and cannot obtain non-global certificates for his or her interest in the securities, except in the special situations we describe below; |
| ∎ | an investor will be an indirect holder and must look to his or her own bank or broker for payments on the securities and protection of his or her legal rights relating to the securities, as we describe above; |
23
Table of Contents
| ∎ | an investor may not be able to sell interests in the securities to some insurance companies and to other institutions that are required by law to own their securities in non-book-entry form; |
| ∎ | an investor may not be able to pledge his or her interest in a global security in circumstances where certificates representing the securities must be delivered to the lender or other beneficiary of the pledge in order for the pledge to be effective; |
| ∎ | the depositary’s policies, which may change from time to time, will govern payments, transfers, exchanges and other matters relating to an investor’s interest in a global security; |
| ∎ | we and any applicable trustee have no responsibility for any aspect of the depositary’s actions or for its records of ownership interests in a global security, nor do we or any applicable trustee supervise the depositary in any way; |
| ∎ | the depositary may, and we understand that DTC will, require that those who purchase and sell interests in a global security within its book-entry system use immediately available funds, and your broker or bank may require you to do so as well; and |
| ∎ | financial institutions that participate in the depositary’s book-entry system, and through which an investor holds its interest in a global security, may also have their own policies affecting payments, notices and other matters relating to the securities. |
There may be more than one financial intermediary in the chain of ownership for an investor. We do not monitor and are not responsible for the actions of any of those intermediaries.
Special Situations When a Global Security Will Be Terminated
In a few special situations described below, the global security will terminate and interests in it will be exchanged for physical certificates representing those interests. After that exchange, the choice of whether to hold securities directly or in street name will be up to the investor. Investors must consult their own banks or brokers to find out how to have their interests in securities transferred to their own name, so that they will be direct holders. We have described the rights of holders and street name investors above.
Unless we provide otherwise in the applicable prospectus supplement, the global security will terminate when the following special situations occur:
| ∎ | if the depositary notifies us that it is unwilling, unable or no longer qualified to continue as depositary for that global security and we do not appoint another institution to act as depositary within 90 days; |
| ∎ | if we notify any applicable trustee that we wish to terminate that global security; or |
| ∎ | if an event of default has occurred with regard to securities represented by that global security and has not been cured or waived. |
The applicable prospectus supplement may also list additional situations for terminating a global security that would apply only to the particular series of securities covered by the applicable prospectus supplement. When a global security terminates, the depositary, and not we or any applicable trustee, is responsible for deciding the names of the institutions that will be the initial direct holders.
24
Table of Contents
We may sell the securities from time to time pursuant to underwritten public offerings, direct sales to the public, negotiated transactions, block trades or a combination of these methods. We may sell the securities to or through underwriters or dealers, through agents, or directly to one or more purchasers. We may distribute securities from time to time in one or more transactions:
| ∎ | at a fixed price or prices, which may be changed; |
| ∎ | at market prices prevailing at the time of sale; |
| ∎ | at prices related to such prevailing market prices; or |
| ∎ | at negotiated prices. |
A prospectus supplement or supplements (and any related free writing prospectus that we may authorize to be provided to you) will describe the terms of the offering of the securities, including, to the extent applicable:
| ∎ | the name or names of the underwriters, if any; |
| ∎ | the purchase price of the securities or other consideration therefor, and the proceeds, if any, we will receive from the sale; |
| ∎ | any options under which underwriters may purchase additional securities from us; |
| ∎ | any agency fees or underwriting discounts and other items constituting agents’ or underwriters’ compensation; |
| ∎ | any public offering price; |
| ∎ | any discounts or concessions allowed or reallowed or paid to dealers; and |
| ∎ | any securities exchange or market on which the securities may be listed. |
Only underwriters named in the prospectus supplement will be underwriters of the securities offered by the prospectus supplement.
If underwriters are used in the sale, they will acquire the securities for their own account and may resell the securities from time to time in one or more transactions at a fixed public offering price or at varying prices determined at the time of sale. The obligations of the underwriters to purchase the securities will be subject to the conditions set forth in the applicable underwriting agreement. We may offer the securities to the public through underwriting syndicates represented by managing underwriters or by underwriters without a syndicate. Subject to certain conditions, the underwriters will be obligated to purchase all of the securities offered by the prospectus supplement, other than securities covered by any option to purchase additional securities from us. Any public offering price and any discounts or concessions allowed or reallowed or paid to dealers may change from time to time. We may use underwriters with whom we have a material relationship. We will describe in the prospectus supplement, naming the underwriter, the nature of any such relationship.
We may sell securities directly or through agents we designate from time to time. We will name any agent involved in the offering and sale of securities and we will describe any commissions we will pay the agent in the prospectus supplement. Unless the prospectus supplement states otherwise, our agent will act on a best-efforts basis for the period of its appointment.
We may authorize agents or underwriters to solicit offers by certain types of institutional investors to purchase securities from us at the public offering price set forth in the prospectus supplement pursuant to delayed delivery contracts providing for payment and delivery on a specified date in the future. We will describe the conditions to these contracts and the commissions we must pay for solicitation of these contracts in the prospectus supplement.
We may provide agents and underwriters with indemnification against civil liabilities, including liabilities under the Securities Act, or contribution with respect to payments that the agents or underwriters may make with respect to these liabilities. Agents and underwriters may engage in transactions with, or perform services for, us in the ordinary course of business.
25
Table of Contents
All securities we may offer, other than common stock, will be new issues of securities with no established trading market. Any underwriters may make a market in these securities, but will not be obligated to do so and may discontinue any market making at any time without notice. We cannot guarantee the liquidity of the trading markets for any securities.
Any underwriter may engage in over-allotment, stabilizing transactions, short-covering transactions and penalty bids in accordance with Regulation M under the Exchange Act. Over-allotment involves sales in excess of the offering size, which create a short position. Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specified maximum price. Syndicate-covering or other short-covering transactions involve purchases of the securities, either through exercise of the over-allotment option or in the open market after the distribution is completed, to cover short positions. Penalty bids permit the underwriters to reclaim a selling concession from a dealer when the securities originally sold by the dealer are purchased in a stabilizing or covering transaction to cover short positions. Those activities may cause the price of the securities to be higher than it would otherwise be. If commenced, the underwriters may discontinue any of the activities at any time.
Any underwriters or agents that are qualified market makers on The Nasdaq Global Select Market may engage in passive market making transactions in the common stock on The Nasdaq Global Select Market in accordance with Regulation M under the Exchange Act, during the business day prior to the pricing of the offering, before the commencement of offers or sales of the common stock. Passive market makers must comply with applicable volume and price limitations and must be identified as passive market makers. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for such security; if all independent bids are lowered below the passive market maker’s bid, however, the passive market maker’s bid must then be lowered when certain purchase limits are exceeded. Passive market making may stabilize the market price of the securities at a level above that which might otherwise prevail in the open market and, if commenced, may be discontinued at any time.
26
Table of Contents
Cooley LLP will pass upon the validity of the securities offered by this prospectus, and any supplement thereto, unless otherwise indicated in the applicable prospectus supplement. GC&H Investments, LLC, an entity that includes current and former partners and associates of Cooley LLP, beneficially owns 10,000 shares of our common stock. Additional legal matters may be passed upon for us or any underwriters, dealers or agents by counsel that we will name in the applicable prospectus supplement.
Ernst & Young LLP, independent registered public accounting firm, has audited our consolidated financial statements included in our Annual Report on Form 10-K for the year ended December 31, 2019, as set forth in their report, which is incorporated by reference in this prospectus and elsewhere in this registration statement. Our consolidated financial statements are incorporated by reference in reliance on Ernst & Young LLP’s report, given on their authority as experts in accounting and auditing.
WHERE YOU CAN FIND ADDITIONAL INFORMATION
This prospectus is part of the registration statement on Form S-3 we filed with the SEC under the Securities Act and does not contain all the information set forth in the registration statement. Whenever a reference is made in this prospectus to any of our contracts, agreements or other documents, the reference may not be complete and you should refer to the exhibits that are a part of the registration statement or the exhibits to the reports or other documents incorporated by reference into this prospectus for a copy of such contract, agreement or other document. Because we are subject to the information and reporting requirements of the Exchange Act, we file annual, quarterly and current reports, proxy statements and other information with the SEC. Our SEC filings are available to the public over the Internet at the SEC’s website at http://www.sec.gov.
INCORPORATION OF CERTAIN INFORMATION BY REFERENCE
The SEC allows us to incorporate by reference information from other documents that we file with it, which means that we can disclose important information to you by referring you to those documents. The information incorporated by reference is considered to be part of this prospectus. Information in this prospectus supersedes information incorporated by reference that we filed with the SEC prior to the date of this prospectus, while information that we file later with the SEC will automatically update and supersede the information in this prospectus. We incorporate by reference into this prospectus and the registration statement of which this prospectus is a part the information or documents listed below that we have filed with the SEC (Commission File No. 001-38853):
| ∎ | our Annual Report on Form 10-K, for the year ended December 31, 2019, filed with the SEC on March 17, 2020; |
| ∎ | our Definitive Proxy Statement on Schedule 14A, filed with the SEC on April 8, 2020 (excluding those portions that are not incorporated by reference into our Annual Report on Form 10-K for the year ended December 31, 2019); |
| ∎ | our Quarterly Report on Form 10-Q, for the quarter ended March 31, 2020, filed with the SEC on May 13, 2020; |
| ∎ | our Current Reports on Form 8-K filed with the SEC on February 13, 2020, February 24, 2020, March 9, 2020 and May 27, 2020; and |
| ∎ | the description of our common stock set forth in our registration statement on Form 8-A, filed with the SEC on March 29, 2019, including any amendment or report filed for the purpose of updating such description. |
27
Table of Contents
We also incorporate by reference any future filings (other than current reports furnished under Item 2.02 or Item 7.01 of Form 8-K and exhibits filed on such form that are related to such items unless such Form 8-K expressly provides to the contrary) made with the SEC pursuant to Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act, including those made after the date of the initial filing of the registration statement of which this prospectus is a part and prior to effectiveness of such registration statement, until we file a post-effective amendment that indicates the termination of the offering of the securities made by this prospectus, which will become a part of this prospectus from the date that such documents are filed with the SEC. Information in such future filings updates and supplements the information provided in this prospectus. Any statements in any such future filings will automatically be deemed to modify and supersede any information in any document we previously filed with the SEC that is incorporated or deemed to be incorporated herein by reference to the extent that statements in the later-filed document modify or replace such earlier statements. We will furnish without charge to each person, including any beneficial owner, to whom a prospectus is delivered, upon written or oral request, a copy of any or all of the documents incorporated by reference into this prospectus but not delivered with the prospectus, including exhibits that are specifically incorporated by reference into such documents. You should direct any requests for documents to:
NGM Biopharmaceuticals, Inc.
333 Oyster Point Boulevard
South San Francisco, California 94080
(650) 392-1768
Attn: Secretary
28
Table of Contents
4,629,630 Shares
NGM Biopharmaceuticals, Inc.
Common Stock
PROSPECTUS SUPPLEMENT
| Goldman Sachs & Co. LLC | Citigroup | Cowen |
| Raymond James | B. Riley Securities |